Chronic inflammatory demyelinating polyneuropathy (CIDP) is a peripheral nerve disease that is autoimmune in nature. Experts talk about a group of diseases united by a common name, which received official recognition only in the 80s of the 20th century. Chronic inflammatory demyelinating polyneuropathy occurs in any age group, but the maximum incidence occurs in adults, more often in middle-aged men. Statistics show that in patients over 50 years of age, the disease has more severe forms that respond less well to treatment. The frequency in adults is 2 cases per 100 thousand population, in children – 1 case per 100 thousand people.
At CELT you can get advice from a neurologist.
- Initial consultation – 4,000
- Repeated consultation – 2,500
Make an appointment
Why does inflammatory demyelination occur?
The causes of CIDP are still being studied; much remains unclear. In more than a third of adult patients, CIDP develops following a viral infection. In children, there is a connection with respiratory infections and age-related vaccinations. Pregnant women in the third trimester are also at risk. However, in half of the patients no clear cause can be found.
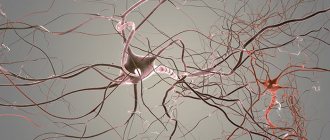
Researchers believe that demyelinating inflammation is triggered by T cells, which begin the destruction of peripheral myelin by producing antibodies. In this case, an inflammatory infiltrate and swelling can occur on any part of the nerve, this also applies to the spinal roots. Therefore, it is sometimes legitimate to talk about polyneuroradiculopathy.
Manifestations
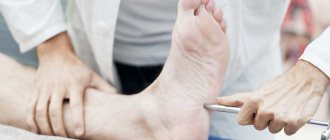
The insidiousness of the disease is that the first symptoms have no specificity and do not cause concern for either the patient or the doctor - everything develops too gradually. For the first few months, there may be no clinical manifestations, although the process is actively underway.
For some reason, the immune system perceives the cells of its body as foreign and begins to actively fight them, forming immune complexes. These complexes literally “eat” the myelin sheath, thereby disrupting the conduction of impulses along the peripheral nerve. In many cases, a hereditary predisposition and familial forms of the disease can be traced.
Sometimes provocateurs can be illness, heavy physical activity, changes in hormonal status, emotional stress, and stress.
There are two main clinical manifestations:
- Weakness or loss of sensation in the arms and/or legs, which is combined with movement disorders. Weakness progresses stepwise and steadily, but slowly. The duration of symptom progression is at least 2 months. Concomitant cranial nerve involvement is possible.
- Decreased tendon reflexes from the limbs to the point of their absence.
Most often, weakness begins in the feet and gradually moves up. In the future, small and precise movements of the hands may be impaired. Muscle atrophy develops much later, or does not occur at all. Decreased sensitivity in the legs leads to instability when walking and pain.
The typical form of CIPD is characterized by symmetry of the lesion, a gradual increase in symptoms, and a good response to treatment. Atypical may be asymmetric or focal, when initially only one plexus or 1 peripheral nerve is involved.
Chronic inflammatory demyelinating polyneuropathy (CIDP) (patient information)
Chronic inflammatory demyelinating polyneuropathy (CIDP)
What do these terms mean?
“Chronic” means that the disease has a long course, and symptoms can steadily progress and recur. To make a diagnosis of chronic polyneuropathy, more than 8 weeks must pass from the onset of the first symptoms. “Inflammatory” implies “inflammation” as the main mechanism of damage to peripheral nerves due to disruption of the complex functioning of the immune system, so this disease can also be called “autoimmune”. “Demyelinating” characterizes the type of damage to peripheral nerves in which the myelin sheath of the nerve is predominantly affected. “Polyradiculoneuropathy” means that the pathological process involves more than one nerve, as well as the roots of the spinal cord and the trunks of the plexuses.
How common is this disease?
CIDP is a fairly rare disease. The average prevalence of patients with CIDP in the world averages up to 0.81-1.90 cases per 100,000 people. Men get sick slightly more often than women. CIDP can debut at any age, even in childhood, but the peak incidence occurs in middle age - 40-50 years.
What are the causes of the disease?
The causes of CIDP are still not fully understood. However, the results of numerous studies and the effectiveness of immunomodulatory therapy point to disruption of the immune system as a key cause of the development of the disease. The immune system is a very complex and harmonious mechanism. The key components of the immune system are antibodies, a number of serum proteins and white blood cells, leukocytes. Normally, the immune system fights foreign agents (proteins, viruses, bacteria). However, in a number of diseases (autoimmune diseases), components of the immune system mistakenly begin to work against their own body. CIDP is one of these diseases. In this case, a reaction develops against the components of the peripheral nerve sheath, which manifests itself in the form of specific symptoms characteristic of CIDP.
Are there risk factors for the disease?
Respiratory viral diseases, surgical interventions, pregnancy, vaccination and other reasons have been described as trigger factors, however, the direct connection of the above factors with the development of the disease has not yet been proven.
Is it possible to transmit the disease by inheritance?
No. There are a number of studies that have identified genes suggested to be involved in the development of the disease. However, their participation in the risk of developing the disease in offspring has not yet been proven.
How does CIDP manifest and what are the features of this disease?
CIDP is a peripheral nerve disease. In the overwhelming majority, the pathological process involves the so-called “thick” nerve fibers, which have a thick layer of myelin sheath, which is the target for this disease. These fibers carry information from the brain and spinal cord to our musculoskeletal system about the motor command and in the opposite direction about the position of the body in space to ensure balance. Therefore, the most common symptoms are:
- weakness in arms and legs
- unsteadiness when walking
- numbness in the hands and feet
- muscle loss and decreased tone
The exact mechanism of CIDP is not fully understood due to the complexity of immune reactions, and therefore the symptoms and nature of the disease may vary. From here, the so-called atypical forms of CIDP are identified, which are somewhat different from the classical course of the disease and may have features of prognosis and treatment. Diagnosis of such forms can be difficult. Atypical forms of CIDP include:
- multifocal form of CIDP (Lewis-Sumner syndrome)
- distal form of CIDP
- purely sensory or purely motor forms of CIDP
- CIDP with acute onset
- chronic immune sensory polyradiculoneuropathy
The course of the disease can also be variable - some patients may develop severe symptoms leading to disability, while others may have minimal neurological impairment; a number of patients may experience frequent exacerbations, while there are cases with a single exacerbation in life.
How was I diagnosed with CIDP?
The key to making a diagnosis is a clinical examination by a neurologist. There are criteria for the disease proposed by the European Federation of Neurological Societies in 2010. To confirm the diagnosis and exclude alternative causes of polyneuropathy, a series of laboratory tests, as well as instrumental research methods, are usually performed. Unfortunately, there is still no “gold” diagnostic standard that could accurately indicate CIDP. Therefore, the diagnosis is often made based on the clinical picture and test results.
The key instrumental method for diagnosing CIDP, as well as any polyneuropathy, is electroneuromyography. This is the study of peripheral nerve conduction using short electrical impulses transmitted along the nerve. Stimulation results in a contraction of the muscle innervated by the nerve being tested, which is recorded by the electrode.
If there is insufficient data to establish a diagnosis, additional MRI of the plexuses, analysis of cerebrospinal fluid, ultrasound of peripheral nerves and, in rare cases, nerve biopsy may be performed. One of the criteria for the correctness of the established diagnosis includes improvement of the condition or stopping the progression of the disease against the background of pathogenetic therapy.
What treatment options are there for CIDP?
Up to 80% of patients with CIDP benefit from therapies that modulate the immune system. Based on large studies, the following treatment options have proven effectiveness in the treatment of CIDP:
- glucocorticosteroid drugs
- intravenous human immunoglobulin preparations
- plasmapheresis
None of the currently known drugs cure CIDP; they only help reduce the activity of the disease, prevent further deterioration or exacerbation, and also reduce the severity of symptoms. In addition, the response to treatment may vary between individuals.
Does each treatment method have its own pros and cons, which are discussed by the doctor, taking into account all individual characteristics?
Glucocorticosteroid drugs (prednisolone, methylprednisolone) are prescribed in the form of tablets and infusions. Initially, a high dose is selected based on body weight, which gradually decreases over time. The duration of administration and the size of the maintenance dose depend on the severity of symptoms, the rate of its progression, and the response to treatment. However, to assess the effectiveness of treatment, the duration of therapy should be at least 12 weeks. Despite its proven effectiveness and relatively low cost, treatment with GCS may be associated with a number of side effects - weight gain, nausea, insomnia, irritability, exacerbation of peptic ulcers, increased blood pressure and blood sugar levels, decreased bone density. Therefore, along with the main drug, complex therapy is prescribed to prevent the development of the above-mentioned consequences of treatment.
Human immunoglobulin preparations are similar in effectiveness to glucocorticosteroids, but the latter are much less likely to cause side effects and are therefore safer. Perhaps the main disadvantage of this treatment is its high cost. Human immunoglobulin preparations are obtained by purifying large quantities (>10,000 L) of human plasma (>1000 donors), which makes them expensive. Treatment consists of a monthly course of intravenous administration of the drug. The course usually takes 4-5 days. In the future, the frequency of drug administration may vary depending on its effectiveness. An important point is the choice of drug. It is necessary to pay attention to its main characteristics: the drug must be suitable for high-dose intravenous immunotherapy, the IgG content must be at least 95%, the amounts of IgA and IgM must be traces. In this case, the amount of IgA should be clearly indicated in the instructions, since the development of allergic reactions is associated with this class of immunoglobulins.
A third treatment option is high-volume plasmapheresis. This method of therapy involves collecting plasma with pathogenic antibodies through a catheter and replenishing it with sterile solutions, protein solutions and/or donor plasma. The procedure is repeated about 5 times, usually at intervals every other day. The effect of this treatment lasts for 3-4 weeks. Given its complexity, this method of therapy is not used for long-term treatment and is often useful in cases of rapid and/or severe exacerbation.
In some patients, despite proper treatment, the disease may still progress or be uncontrollable. In these cases, immunosuppressants (mycophenolate mofetil, azathioprine, cyclosporine, cyclophosphamide) or monoclonal antibodies (rituximab) are prescribed. The prescription of these drugs should come from a doctor experienced in their use, taking into account all indications and contraindications, followed by careful monitoring of the effectiveness and safety of therapy.
Do you need to make any changes to your usual lifestyle?
Yes. There are a number of recommendations for patients diagnosed with CIDP:
- avoid any viral and bacterial infections (respiratory viral, enteroviral diseases, etc.)
- move more - in case of severe movement disorders, physical rehabilitation, occupational therapy or walking support aids - orthoses, etc. may be prescribed.
- limit smoking, alcohol consumption, which has a negative effect on blood circulation, aggravating the course of polyneuropathy
- exclude the use of neurotoxic drugs that aggravate the course of polyneuropathy
- Careful foot care is very important, especially if you have concomitant diabetes. It is necessary to inspect your feet daily for cuts, calluses, and ulcers.
- Eat a low-fat diet rich in grains, fruits and vegetables.
- avoid prolonged compression of the limbs
What are the prognosis for this disease?
In general, life expectancy does not differ from that of people who do not have this disease. The course of the disease can be different - occur with frequent exacerbations, have a slowly progressive course, or achieve stable remission with minimal clinical manifestations. Timely administration of treatment, careful monitoring of the patient and the effects of the therapy are extremely important for the prognosis.
If you have symptoms of polyneuropathy or have been diagnosed with “Polyneuropathy” or “CIDP”, you can undergo a comprehensive examination at the Center for Diseases of the Peripheral Nervous System of the Federal State Budgetary Institution Scientific Center for Neuroscience, where they will help you clarify the diagnosis, identify the causes of damage to the peripheral nerves and prescribe therapy based on evidence-based medicine .
Employees of the Center for Peripheral Nervous System Diseases provide consultations to patients on an outpatient basis within the framework of compulsory medical insurance and on a commercial basis.
MAKE AN APPOINTMENT AND ENMG/iEMG BY MULTI-CHANNEL PHONE +7 +7
Diagnostic principles
MRI of the spine
- Cost: 16,000 rub.
More details
Neurologists at the CELT clinic know that the initial manifestations of CIDP occur under the guise of many other common diseases, for which patients undergo long and unsuccessful treatment. Diagnostics includes the following studies:
- Electroneuromyography, which makes it possible to identify disturbances in the conduction of excitation along nerve fibers, and to carry out differential diagnosis with other diseases of the peripheral nervous system.
- Blood test for antibodies.
- MRI of the spinal cord (spine) with contrast enhancement; contrast accumulation sometimes occurs in the area of the inflamed nerve roots.
The results of these studies, as well as the response to therapy, make it possible to distinguish CIDP from other types of polyneuropathies.
Therapy
Approximately 80% of people with CIDP experience an outcome in which the overall condition progresses to a milder form. Taking into account the research conducted in the field of treatment of pathology, the following medications are highly effective:
- glucocorticoids;
- immunoglobulins for intravenous administration.
Along with taking these medications, blood is taken, purified, and returned, or some part, back into the bloodstream.
At the moment, there is no drug that completely cures polyradiculoneuropathy. However, their combined use reduces the progression of the disease, inhibits subsequent deterioration or exacerbation, and also reduces the severity of symptoms.
Our doctors
Novikova Larisa Vaganovna
Neuropathologist, Candidate of Medical Sciences, doctor of the highest category
Experience 39 years
Make an appointment
Pankov Alexander Rostislavovich
Neurologist
40 years of experience
Make an appointment
Treatment rules
Treatment begins with the administration of corticosteroid hormones. The choice of a specific drug and its dosage depends on the specific situation. The duration of the course of treatment is selected individually; a transition to maintenance doses is required. The disease requires a long period of immunosuppression, otherwise exacerbations are inevitable. Hormones are most often administered intravenously.
According to indications, plasmapheresis is used, during which immune complexes circulating in the blood are removed. Plasmapheresis and intravenous immunosuppression are first-line treatments.
Response to specific treatment and high-quality stable remission after it are considered an important diagnostic sign.
Neurologists at the CELT clinic believe that CIDP should be considered in all cases of progressive polyneuropathy. Particularly alarming in this sense is nerve damage in diabetes mellitus, when low glucose levels should not cause severe disorders of muscle strength and sensitivity.
Children who respond to treatment with rapid remission deserve special attention. Their condition must be constantly monitored, since early refusal of treatment leads to another exacerbation, which is always more severe than the initial disorder.
Human immunoglobulins for intravenous administration in the treatment of Guillain–Barré syndrome in children
Definition
Guillain–Barré syndrome is an acute, rapidly progressive autoimmune lesion of the peripheral nervous system, manifested as paresthesia of the limbs, muscle weakness and/or flaccid paralysis (monophasic immune-mediated neuropathy) [1–7]. The described neurological deficit develops as a result of damage to the roots of the spinal cord, spinal and cranial nerve trunks. Guillain–Barré syndrome is a relatively rare disease, with an incidence of 1–1.9 cases per 100,000 people [1–3]. The disease, first described by O. Landry (1859) in the mid-19th century, was named after two French neurologists G. Guillain and JA Barré, who studied it at the beginning of the 20th century. [8, 9]. Less commonly, the disease is called Guillain-Barré-Strohl syndrome or Landry-Guillain-Barré-Strohl syndrome. A differential diagnostic criterion that allows one to distinguish this syndrome from poliomyelitis and other neuropathies is muscle weakness in combination with the so-called albumin-cytological dissociation (increased protein in the cerebrospinal fluid with normal cytosis), which was established by G. Guillain, JA Barré and A. Strohl (1916 ) [9]. For a long period, steroids have been used to treat Guillain–Barré syndrome: adrenocorticotropic hormone (ACTH) drugs, cortisone, dexamethasone, prednisolone and methylprednisolone. This method was proposed in 1952 by JS Stillman and WF Ganong (USA) [10], later it was recognized as insufficiently effective [11]. The first mention of the successful use of plasma exchange in the treatment of Guillain-Barré syndrome (chronic form) belongs to RL Levy et al. (1979) from Great Britain (PubMed electronic database) [12]. The results of the first randomized clinical trial on the use of intravenous human immunoglobulins in the treatment of Guillain–Barré syndrome were published by FG van der Meché and PI Schmitz (Netherlands, 1992) [13].
Clinical variants and classification
In the relatively recent past, only two clinical variants of Guillain-Barré syndrome were considered: acute idiopathic form and chronic (recurrent). It was assumed that the first (main) form accounts for up to 95% of cases of the disease, and the remaining 5% are chronic [1]. To date, it is customary to distinguish at least five varieties (clinical variants) of Guillain-Barré syndrome. These include the following conditions:
- acute inflammatory demyelinating polyneuropathy (AIDPN);
- acute motor-sensory axonal neuropathy (ASAN);
- acute motor axonal neuropathy (AMAN);
- Miller Fisher syndrome;
- chronic inflammatory demyelinating polyneuropathy (CIDP) [1, 13–17].
Note that it is this form of the disease (sporadic variant of Guillain-Barré syndrome) that occurs most often. In addition to the above, there are rare and/or atypical variants of the disease.
Etiology
The etiological factors of Guillain-Barré syndrome have not been fully studied, which allows us to consider the disease as an idiopathic polyneuropathy. However, a number of pathogenic microorganisms can be considered as etiological factors, since infection with them often precedes the development of Guillain-Barré syndrome. These include: cytomegalovirus (CMV), Epstein-Barr virus, Haemophilus influenzae type b, Mycoplasma pneumoniae, Campylobacter jejuni (C. jejuni) and many other causative agents of infectious diseases and processes [1, 2, 4, 18]. It is also impossible to exclude the etiological role of preventive immunization (anti-poliomyelitis, anti-rabies, anti-diphtheria, anti-influenza, etc.) in the development of this severe, rapidly progressing disease [1–7, 14–16, 18]. According to many authors, the disease is a consequence of an abnormal T-cell response induced by an infectious process. Thus, Guillain-Barré syndrome is considered as an acquired immune-mediated neuropathy that develops as a result of an aberrant immune response to a previous immune-activating event (viral infection, vaccination, etc.) [1, 4, 18]. It is noteworthy that C. jejuni is a causally significant infectious agent in a third of patients, and molecular mimicry between gangliosides and liposaccharides (epitopes GM1, GM1b, GD1a, GQ1b, GalNAc-GD1a) of this bacterium promotes the production of anti-ganglioside antibodies. High titers of anti-ganglioside antibodies of the IgM, IgG and IgA classes, which react with epitopes of the axoplasmic section of axons and the myelin sheath, are found in the blood serum of 40% of patients with Guillain-Barré syndrome [1–4, 14, 18].
Pathogenesis
In the classic form of Guillain-Barré syndrome (acute inflammatory demyelinating polyneuropathy), damage to the fibers of motor and sensory neurons occurs. The main structures that are affected pathologically are predominantly the motor neurons of the anterior roots and adjacent proximal plexuses. A characteristic phenomenon is pronounced segmental inflammatory demyelination, which is accompanied by focal and diffuse infiltration of T-lymphoid and monocyte-macrophage cells at all levels of the peripheral nervous system. Inflammatory cells accumulate around small vessels of the endoneurium/epineurium. Complement-mediated binding of antibodies to epitopes located on the surface membrane of Schwann cells precedes T-cell infiltration [1, 4, 19]. Morphological changes in acute and chronic inflammatory demyelinating polyneuropathy resemble disorders in experimental autoimmune neuritis; similar pathological changes are observed in Miller-Fisher syndrome [1, 4, 20]. Axonal variants of Guillain-Barré syndrome (acute motor-axonal and motor-sensory neuropathies) are characterized by the absence of pronounced signs of inflammation and the presence of axonal degeneration of nerve fibers. Changes in the central nervous system (CNS) in these disease variants are secondary to axonal degeneration. In acute motor axonal neuropathy, the motor nodes of Ranvier are primarily susceptible to “immune attack” [1, 4, 7, 14].
Immunopathological reactions lead to autoimmune tissue damage associated with molecular mimicry mechanisms involving superantigens and stimulation of cytokines [1, 4, 20]. The first step in the immunopathogenesis of the disease is the presentation of antigen to naive T cells with their subsequent activation, circulation in the bloodstream and fixation on the venular endothelium of peripheral nerves. T cells then cross the blood-brain barrier, penetrate the endothelial layer into the perivascular region and migrate to the endoneurium, using molecular mechanisms of adhesion (selectins, leukocyte integrins and their counterreceptors). The final step in the pathogenesis of Guillain–Barré syndrome is the entry of T cells and autoantibodies into the endoneurium along with macrophages, where, using antibody-dependent and T-cell mechanisms, autoantigens are identified on axonal or Schwann cells. This leads to severe tissue damage, which is facilitated by active phagocytosis of cells of the monocyte-macrophage line [1, 4, 19]. R. S. Tsang and A. Valdivieso-Garcia (2003), B. C. Kieseier et al. (2004), as well as D. Lambracht-Washington and GI Wolfe (2011) emphasize that cytokines (IL-10, IL-12, IL-18, tumor necrosis factor alpha, leukemia inhibitory factor) and chemokines (attractant protein monocytes, interferon-inducible protein) are actively involved in the inflammatory process in Guillain–Barré syndrome [19–21].
Genetic and immunogenetic aspects of the disease
When studying antigens of the HLA system (major histocompatibility complex), D. Adams et al. (1977) showed that genes associated with the HLA-A and HLA-BB loci do not participate in the development of Guillain–Barré syndrome [22]. Other studies have identified an association between the HLA-54, HLA-CW1, HLA-DQB*3 antigens and Guillain–Barré syndrome [1]. E. E. Magira et al. (2003) They found a positive correlation between acute inflammatory demyelinizing polyneuropathy and the DQB1*0603 allele with a unique epitope dqβed70–71, as well as a negative correlation - with AQB1*0503 alleles, DQB1*0601, DQB1 0602 and DQB1*0603 characterized by epitope RD P55- 57 [23]. All of the above allowed K. Geleijns et al. (2005) concluded that the HLA class may be determining in various variants of Guillain–Barré syndrome, and the disease itself is a complex genetic disorder [24]. Differences in the distribution of HLA-DQB epitopes have been identified in patients with acute inflammatory demyelinating polyneuropathy and acute motor axonal neuropathy (immunogenetic heterogeneity). In turn, earlier M. Koga et al. (1998) found a strong relationship between HLA-DQB1*03 and previous infection with C. jejuni [25]. It is assumed that the detection of C. jejuni DNA in myelomonocytic cells indicates the presentation of neuritogenic antigens to T cells by the HLA class II complex [1, 4]. In their recent work, KH Chang et al. (2012) described 256 genes and 18 gene networks significantly associated with Guillain–Barré syndrome; among them, the most common genes were FOS, PTGS2, HMGB2 and MMP9 [26].
Clinical manifestations of Guillain–Barré syndrome
During Guillain-Barré syndrome, it is customary to distinguish 3 stages:
- progression;
- persistent symptoms
- recovery [1–4, 6, 7, 27, 28].
The first symptoms of Guillain–Barré syndrome, as a rule, appear in the absence of a pronounced increase in temperature [1, 4]. The classic manifestations of the disease are progressive (ascending) paralysis of the muscles of the limbs and respiratory muscles, which is accompanied by sensitivity disorders of the polyneuropathic type; subsequently, patients experience vegetative-trophic disorders [1–7]. In almost all cases, the appearance of symptoms of the disease is preceded by acute respiratory infections, sometimes trauma or surgery, and less often hypothermia [1–3].
Acute inflammatory demyelinating polyneuropathy
The clinical manifestations of ARDP are a direct consequence of disturbances in saltatory conduction along myelinated axons (a so-called conduction block occurs). Notably, approximately two thirds of patients had an episode of acute respiratory infection or gastroenteritis in the 2 weeks before the onset of muscle weakness [1–4]. A characteristic sign of AIDPN is the sudden onset of neurological symptoms. Most patients experience pain (up to 80%) and paresthesia (20%); Quite typical symptoms are ataxia and cranial nerve palsies/palsies. Approximately 30% of children have sphincter dysfunction. Damage to the sympathetic nervous system is manifested by various autonomic disorders (hypertension, postural hypotension, profuse sweating, thermoregulation disorders, etc.) [1–4, 6, 7]. Paralysis of the respiratory muscles is a typical and severe complication of acute inflammatory demyelinating polyneuropathy, requiring mechanical ventilation and/or tracheostomy [1, 14, 29].
Chronic inflammatory demyelinating polyneuropathy
Some patients (from 5 to 10%) who have previously suffered acute inflammatory demyelinating polyneuropathy may experience relapses of the disease (one or more) over a period of 2 months to several years. This circumstance allows us to state that they have chronic inflammatory demyelinating polyneuropathy [1–4, 6, 7, 28, 29]. The factors that initiate disease relapses remain unknown in most cases. It is assumed that they relate to immune-mediated mechanisms and reactions of the body [29].
Acute motor-sensory axonal neuropathy
The symptoms of this form of Guillain–Barré syndrome are practically indistinguishable from the manifestations of acute inflammatory demyelinating polyneuropathy, but the prognosis of the disease is more unfavorable. In children with OMSAN, severe axonal degeneration is observed with primary damage to the axons of motor and sensory nerves. Subsequently, delayed and incomplete recovery of impaired neurological functions is expected (compared to OVDP) [1, 4, 28, 29].
Acute motor axonal neuropathy
OMAN is a variant of the disease in which there is a predominant lesion of the proximal part of motor neurons [1, 6, 29]. In some patients (not in all cases), at the onset of OMAN, hyperthermia (fever), hemorrhagic conjunctivitis, asymmetric muscle weakness, and the presence of pleocytosis in the cerebrospinal fluid are observed. An electroneuromyographic study (ENMG) reveals a decrease in motor action potential in patients, as well as a denervation type of ENMG. At the same time, preservation of the speeds of neuromotor and neurosensory conduction is noted [1, 4].
Miller-Fisher syndrome
A distinctive feature of this form of Guillain–Barré syndrome is the presence of external ophthalmoplegia in combination with ataxia and areflexia, which appears during the first week after the onset of the disease [1, 4, 30]. The first signs of Miller-Fisher syndrome are diplopia, as well as bilateral facial nerve paresis (noted in 50% of patients). Ophthalmoplegia (internal) is recorded in 70–75% of children with Miller-Fisher syndrome [1, 4, 6]. The described symptoms intensify, reaching maximum severity, which persists for 1–2 weeks, after which a gradual recovery of neurological functions is noted (usually it is complete or almost so). In the cerebrospinal fluid, some patients exhibit increased protein content and pleocytosis, and in rare cases, typical albuminocytological dissociation (as in AFDP) [1–4]. An ENMG study in children with Miller-Fisher syndrome allows, in some cases, to register a slowdown in conduction along the motor and sensory nerves [29, 30].
Forecast
Most patients with Guillain–Barré syndrome experience spontaneous recovery, although up to a quarter of patients may require mechanical ventilation (MV). On average, the duration of the disease progression stage is about 4 weeks, and the stage of persistent symptoms is about 2 weeks. Active restoration of lost neurological functions continues for about a month and a half; subsequently, the pace of recovery slows down significantly. It is believed that neurological disorders that persist after the end of the acute period of the disease are subsequently observed in 7–22% of children (in adult patients - in 20–30% of cases). Relapse/relapses of the disease (chronic inflammatory demyelinating polyneuropathy) occur in 3–10% of patients. Mortality in Guillain–Barré syndrome can reach 10%, although more often it is recorded at the level of 2–5% [1, 4, 28, 31].
Diagnostics
The main diagnostic criteria for Guillain–Barré syndrome are: 1) progressive motor weakness involving more than one limb in the pathological process; 2) areflexia or severe hyporeflexia; 3) the content of no more than 50 monocytes and/or 2 granulocytes in 1 μl of cerebrospinal fluid [1, 4, 27]. Additional signs confirming this diagnosis include: 1) absence of hyperthermia (fever) at the onset of the disease; 2) the beginning of restoration of neurological functions 2–4 weeks after the cessation of progression; 3) relatively symmetrical muscle weakness; 4) moderate signs of sensory impairment; 5) symptoms of damage to the cranial nerves (CN); 6) an increase in protein content in the cerebrospinal fluid after 1 week after the onset of characteristic symptoms of the disease; 7) slowing of nerve conduction velocity or prolongation of F-waves (in ENMG study); 7) the presence of autonomic (autonomous) dysfunction [1, 4, 27, 32, 33]. An ENMG study of the affected muscles in Guillain-Barre syndrome reveals a denervation type of curve [32, 33]. The diagnostic criteria confirming the presence of Guillain-Barré syndrome include the following 3 indicators determined by analyzing the cerebrospinal fluid: 1) increased protein content; 2) increase in albumin fraction; 3) absence of increased cytosis [1].
Differential diagnosis
Guillain-Barré syndrome must be differentiated from conditions such as acute (epidemic) polio, botulism, myasthenia gravis, acute myelitis, diphtheria polyneuropathy, acute polyneuropathy of other origins, etc. [1–7, 13].
Therapy
A mandatory condition for the treatment of Guillain-Barré syndrome is urgent hospitalization in the intensive care unit (due to the potential need for mechanical ventilation) [1, 34]. Among non-pharmacological methods of treating Guillain-Barré syndrome, plasmapheresis occupies a special place as the only method of instrumental (extracorporeal) treatment of the disease [35, 36]. JC Raphael et al. (2002) indicate that plasmapheresis is the first treatment method that has advantages over supportive care (with proven effectiveness) [35]. Among the known and previously used methods of drug therapy for the disease, such as immunosuppressants are cyclosporine, cyclophosphamide and azathioprine; corticosteroids; beta-interferon preparations; autologous stem cell transplantation; human immunoglobulins for intravenous administration, only the latter can be considered effective from the standpoint of evidence-based medicine [36]. This is evidenced by data from a systematic review presented by RA Hughes et al. (2010) [37]. The effectiveness of other methods of pharmacological treatment of Guillain–Barré syndrome seems questionable at best [38–41]. There are 2 main regimens for prescribing human immunoglobulins for intravenous administration to children with Guillain-Barré syndrome: 1) 0.4 g/kg/day for 5 days; 2) 2.0 g/kg once [4, 5, 13, 20, 34–36]. The first mode is more often used and is considered preferable.
In the treatment of Guillain-Barré syndrome, both human immunoglobulins for intravenous administration containing only IgG (Intratect) and intravenous immunoglobulins enriched with IgM (Pentaglobin), produced by Biotest Pharma GmbH (Germany), can be successfully used. The latter should be given preference, since IgM stimulates remyelination to a greater extent due to interaction with antigens of the central nervous system and oligodendrocytes. In turn, IgG also stimulates remyelination mainly due to immunomodulatory activity (it also penetrates the blood-brain barrier better). Thus, when using human immunoglobulins for intravenous administration containing IgM, an additional stimulating effect on the remyelination process is achieved. When using Pentaglobin, containing 50 mg of human plasma proteins in 1 ml (of which IgG - 38 mg, IgA and IgM - 6 mg each), in the treatment of Guillain-Barre syndrome, it is proposed to dose the drug at the rate of 0.4 g / kg / day (i.e. 8 ml/kg/day) [42]. Human immunoglobulin for intravenous administration Intratect, 1 ml of which also contains 50 mg of human plasma proteins, of which IgG is at least 96% (subclasses IgG1 - 57%, IgG2 - 37%, IgG3 - 3%, IgG4 - 3%), dosed similarly [42].
1 ml of the drug Intratect, used in the treatment of Guillain-Barré syndrome, contains 50 mg of human plasma proteins, of which IgG is at least 96% (subclasses IgG1 - 57%, IgG2 - 37%, IgG3 - 3%, IgG4 - 3%) , it is recommended to dose the drug at the rate of 0.4 g/kg/day (i.e. 8 ml/kg/day) [42]. It should be noted that in the production of Intratek, an innovative technology is used - 2-stage cation exchange chromatography, which makes it possible to obtain a highly purified IgG preparation and ensure its exceptionally good tolerance by patients. According to the above-mentioned systematic review by RA Hughes et al. (2010), immunotherapy with human immunoglobulins for acute demyelinating polyneuropathy promotes rapid restoration of neurological functions, at least as effective as plasmapheresis [36]. AK Meena et al. (2011) give preference to human immunoglobulins for intravenous administration, emphasizing the high efficiency of such immunotherapy, as well as the small number of side effects associated with their use [34].
Scientific research in the field of studying the problem of Guillain-Barré syndrome continues, as evidenced, in particular, by the recent publication by YZ Wang et al. (2012) from China, who studied the expression of Toll-like receptors (TLRs) 2, 4 and 9 in patients with this pathology. Researchers found not only an increase in TLR2, TLR4 and TLR9 (in combination with associated signaling molecules) in patients with Guillain–Barré syndrome compared to healthy donors, but also a significant positive correlation between high levels of these indicators and the severity of neurological deficits [45]. Since TLRs play an important role in activating the cellular immune response, the use of intravenous human immunoglobulins may have a positive effect on the described immunity parameters in Guillain-Barré syndrome.
N.B.
Human immunoglobulins for intravenous administration, which have been included in the arsenal of pharmacological agents of doctors of various specialties since the 1980s, have been successfully used in the treatment of diseases with established or suspected autoimmune origin. The mechanisms of the therapeutic action of human immunoglobulins for intravenous administration have not been fully elucidated, but it is known that they have the following properties: anti-inflammatory; the ability to nonspecifically stimulate T-lymphocytes; to neutralize antiviral, antibacterial and autoantibodies; the ability to bind activating complement components and neutralize activated complement [43]. It is assumed that when intravenous immunoglobulins are used in the treatment of Guillain–Barre syndrome, there is a provision of anti-idiotypic antibodies with selective suppression of anti-myelin antibodies [1, 43]. It should be remembered that intravenous human immunoglobulins have been used for a number of years in the treatment of other demyelinating diseases of the nervous system, in particular multiple sclerosis [44].
Prevention options
Prevention of CIDP is only possible secondary, since a person cannot change his genetic code or avoid infection with viruses. In all conditions where weakness and sensory disturbances occur in the limbs, a thorough clinical examination is required.
CELT neurologists are always wary of such manifestations and make maximum use of diagnostic capabilities. Effective measures to prevent relapses and exacerbations, and reduce the severity of disease progression include complete completion of the treatment course prescribed by the doctor. You should not stop when minimal improvement has been achieved. Treatment must be carried out to the end and appear on time for repeated examinations by the attending physician.
Neurologists at the CELT multidisciplinary clinic have extensive practical experience and are constantly expanding their knowledge and are able to recognize complex forms of demyelinating inflammation.
Make an appointment through the application or by calling +7 +7 We work every day:
- Monday—Friday: 8.00—20.00
- Saturday: 8.00–18.00
- Sunday is a day off
The nearest metro and MCC stations to the clinic:
- Highway of Enthusiasts or Perovo
- Partisan
- Enthusiast Highway
Driving directions