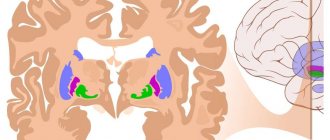
Polymicrogyria, agyria and pachygyria are congenital malformations of the brain, also called brain malformations.
These anomalies arise as a result of a disrupted process of formation of the brain or its individual structures during the prenatal period, and represent abnormal changes in the structure of brain structures.
There are no exact data on the prevalence of congenital brain defects, but polymicrogyria is considered the most common.
More details about each malformation:
- Polymicrogyria is a rare pathology of the development of the cerebral cortex, in which a significant number of small and underdeveloped convolutions are formed on the surface of the cerebral hemispheres. It is often combined with other genetic pathologies and abnormalities in the development of the cerebral cortex. Most often, pathology develops in the area of the Sylvian fissure (60% of cases), but can occur in any part of the cerebral cortex. Pathology can be diffuse, multifocal and focal. It can also be divided into unilateral (40% of cases) and bilateral (60% of cases), symmetrical and asymmetrical.
- Agyria (lissencephaly) is an anomaly in the formation of the cerebral cortex, in which the gyri are underdeveloped, weakly expressed, or absent, and the architecture of the cortex is disrupted. The appearance of the child's brain is identical to the appearance of the fetal brain at 3-4 months of the prenatal period. Agyria is a severe form of lissencephaly. It can be either an idiopathic disease or accompany other pathologies (Miller-Diecker syndrome, Norman-Roberts syndrome, Walker-Warburg syndrome, Fukuyama congenital muscular dystrophy).
- Pachygyria is a rare developmental anomaly of the central nervous system, in which a small number of wide and flat convolutions are formed in the cerebral cortex. With pachygyria, the main gyri are enlarged, and the secondary and tertiary gyri are absent, the sulci are shortened and straightened, and the architecture of the cerebral cortex is disrupted. This pathology is considered an “incomplete”, milder form of lissencephaly.
Brain malformations: polymicrogyria, agyria and pachygyria
Polymicrogyria, agyria and pachygyria are congenital malformations of the brain, also called brain malformations.
These anomalies arise as a result of a disrupted process of formation of the brain or its individual structures during the prenatal period, and represent abnormal changes in the structure of brain structures. There are no exact data on the prevalence of congenital brain defects, but polymicrogyria is considered the most common.
More details about each malformation:
Definition
If the migration of neurons is disrupted, the cortex ceases to normally form convolutions and grooves. Its multilayer structure is disrupted. Lissencephaly is a congenital malformation of the child’s cerebral cortex, in which a thin, four-layer cortex is formed and there are no gyri or sulci. The complete absence of gyri is called agyria, and this is the most severe form of lissencephaly. In the event that the gyri exist, but are severely deformed and underdeveloped, this condition is called pachygyria. The first photo shows agyria, the next photo compares the gyri and sulci of the cerebral cortex of a healthy child and a patient with lissencephaly.
Lissencephaly and agyria can be an independent defect in the development of the cortex in a child, or accompany such congenital diseases as Miller-Diecker and Norman-Roberts syndromes, which are also rare pathologies.
Provoking factors
The main reason for the development of polymicrogyria, pachygyria and agyria is the incorrect process of neuronal migration, which is caused by:
Polymicrogyria can be caused by cytomegalovirus infection, insufficient oxygen saturation of the placenta, and mutations occur in the genes COL18A1 (21q22.3), PAX6 (11p13), K1AA1279 (10q22.1), TUBB2B (6p25), EOMES (3p21.3-p21.2 ), RAB3GAP1 (2q21.3), SRPX2 (Xq21.33-q23).
Agyria can occur due to viral infections that penetrate the uterus or fetus during the first trimester, insufficient blood supply to the fetal brain in the first months of pregnancy. Among the genetic factors are mutations in the RELN gene (7q22), genes of the X chromosome, and 17 chromosomes.
In pachygyria, mutations occur in the LIS1 (17p13.3) and DCX (Xq22.3-q23) genes.
Diagnosis of the disease
The primary diagnosis of lissencephaly is not particularly difficult. For this, the methods that exist in every major city are used:
- Ultrasound of the fetal brain at the age of 22-27 weeks, after the main process of formation of grooves and convolutions;
- computed tomography, which allows you to quickly determine the main variants of the topography of the gray and white matter of the brain;
- Using MRI, you can determine the type of disease and make a presumptive diagnosis of genetic disorders;
- Electroencephalography (EEG), with which a child can obtain a characteristic picture of the electrical activity of the malformed cerebral cortex.
Characteristic symptoms
The following symptoms are characteristic of polymicrogyria:
Pachygyria manifests itself before the age of 2 years. Its main features are:
Agyria manifests itself by 3-5 months of a child’s life, less often by 8-9 months; the main symptom of the pathology is expressed in the rapid lag of the child in physical and mental development.
The anomaly may also manifest itself with the following symptoms:
Cross-sectional photo of the brain and MRI of a child diagnosed with agyria
How to recognize
In some cases, a brain malformation can be recognized in utero, during an ultrasound scan of the fetus. During pregnancy, the formation of fissures and convolutions in the fetus can be assessed no earlier than the 20th week, since in the early stages of pregnancy the smooth brain is a normal stage of fetal development. The problem can also be noticed when performing an ultrasound of the brain (neurosonography) in the first months of a child’s life. At birth, brain problems in a child may be indicated by malformations of the fingers, structural features of the head and face, heart defects, etc.
Article on the topic
The brain is on its own. What kind of “creature” lives in our skull? But it also happens that with a favorable course of pregnancy and childbirth, a child with such a pathology is regarded as healthy and does not cause concern among parents at first. However, developmental delay gradually becomes noticeable, which is often the first complaint. During examination, in most cases, the doctor pays attention to the reduced size of the head.
Later, epileptic seizures occur, and sometimes parents consult a doctor due to their occurrence. The onset of epileptic seizures is noted when the child reaches the age of 3-5 months, less often they begin after 9 months. The first type of seizures are epileptic spasms. Such attacks are controlled with special medications. To select them correctly for a child, a number of studies should be carried out: EEG, video-EEG monitoring with sleep.
A child with such a pathology will further lag behind in development (both mental and physical). Epileptic seizures will become increasingly difficult to stop and treat. In the most severe cases, the child does not acquire practically any skills (does not hold his head up, does not roll over, does not sit, etc., there is no speech). Paresis and paralysis of the limbs are formed (as a rule, with an increase in muscle tone). Disturbances in the functioning of internal organs are also possible (malformations of the heart, kidneys, gastrointestinal tract, reproductive system, finger abnormalities and cleft palate). For example, such a severe picture is characteristic of lissencephaly in Miller-Dieker syndrome and some other forms of the disease.
"Autopsy" of the brain. Will intelligence drugs divide people into gods and fools? More details
Establishing diagnosis
The following studies are used to diagnose brain malformations:
With polymicrogyria, the cerebral cortex is abnormally thin, and the subarachnoid spaces and ventricles of the brain are enlarged. Defeats
can be located on one or both sides, can be local or extensive. The cortex may appear abnormally thick due to the large number of small convolutions that coalesce to form thick, deep masses.
Pachygyria is determined by MRI results, which show thickening of the cerebral cortex, small gyri, and an underdeveloped Sylvian fissure.
In the prenatal period, it is possible to determine brain malformations using fetal ultrasound. If developmental abnormalities are suspected, they are referred for additional studies (MRI, genetic analysis of candidate genes).
Prevention
The only thing that can somehow prevent the development of the problem is careful pregnancy planning. Future parents should definitely undergo a genetic consultation to eliminate the risk of mutations. It is also worth getting tested for TORCH infections.
When a child with lissencephaly appears in a family, a genetic examination when planning further children is necessary, since from the first descriptions of the disease there have been cases of its detection in several children in the same family. In these cases, it is especially important to conduct genetic studies on a sick child. A geneticist will tell you which ones exactly. After all the research, you will need to consult a doctor and decide on the safe planning of offspring.
Medicine is practically powerless
There are no effective methods for treating brain malformations in modern medicine. Therapy is aimed at relieving symptoms and maintaining the functioning of the body at an acceptable level.
For polymicrogyria and pachygyria, the following methods are recommended to alleviate the condition of patients:
For agyria, classical methods of therapy are considered ineffective: taking nootropic drugs, massage, physiotherapy.
Treatment with anticonvulsants and centrally acting muscle relaxants, which reduce tone in skeletal muscles, shows the greatest effectiveness.
Often epilepsy, which accompanies pathologies of brain development, turns out to be resistant to therapy, and then it is recommended to use a combination of two drugs.
Research is being conducted to treat these anomalies using stem cell transplantation and growth factor transplantation.
Prognosis and life expectancy
Patients with polymicrogyria who have mild forms of the pathology can survive into adulthood. In severe forms, death occurs at an early and young age due to pneumonia or seizures.
There is no single prognosis for patients with pachygyria. It can vary in each specific case and depends on the severity of the brain pathology and the intensity of the symptoms. But usually children with pachygyria live no longer than 20 years.
Underdevelopment of the brain in children with agyria is irreversible and the prognosis is unfavorable. Most patients do not survive beyond the age of 5 years. All patients have complete disability.
Due to depressed swallowing reflexes, tube feeding is often required. Death can occur due to diseases that occur against the background of the underlying pathology: intestinal paresis, cardiovascular failure, respiratory failure, atrophy of the respiratory muscles, hypostatic pneumonia.
Source
Pachygyria brain symptoms
Terms microgyria
and
polymicrogyria
(PMG) describe pathological changes in the cerebral cortex, represented by many short, shallow gyri, forming a pathological pattern that can vary macroscopically. In some cases, when assessing the surface of the brain, the broad gyri are difficult to distinguish from areas of pachygyria, but the cortical ribbon has a typical polymicrogyric appearance with fusion of molecular layers (Friede, 1989, Harding and Sorr, 1997). Microgyria can be generalized, unilateral, multifocal and focal.
There are two main histological types of polymicrogyria (PMG), but transitional or mixed forms often occur. The most common unconditioned developmental disorder is microgyria with undivided layers (Ferrer, 1984, Harding and Sorr, 1997), in which there is a single wavy cell layer between the white matter and the molecular layer.
In classic four-layer microgyria, the first layer of small pyramidal cells, located below the molecular layer, is separated by a low cellular layer from a deep layer containing large pyramidal cells. The low cellular layer corresponds to normal layers IV and V, with which it merges at the boundaries of the microgyric cortex (Williams and Caviness, 1984, Evrard et al., 1989). This phenomenon allows us to suggest that post-migration disorders occur in the fifth or sixth month of pregnancy with secondary laminar necrosis developing as a result of circulatory failure and hypoxia. This interpretation is supported by the fact that classical PMG develops due to pathological events occurring after the middle of the current pregnancy, such as severe trauma, carbon monoxide toxicity or maternal asphyxia (Larroche, 1986, Barth, 2003b), and only in one of the monozygotic twins (Sugama and Kusano, 1994). Evrard et al. (1989) identified two peak periods for circulatory failure: between 20 and 24 weeks and during the last 10 weeks of gestation.
Insufficient blood flow may also be responsible for microgyria in fetal infections (Marques-Dias et al., 1984), which can cause vascular damage (Barkovich and Lindan, 1994) or circulatory failure, as well as porencephalic defects. However, individual PMGs can form shortly before the end of the migration processes and arise as a result of the death of the surface layers, damaging the glial conductors before the late migrating cells complete the migration process (Dvorak et al., 1978). In these cases, neurons arrested in the process of migration are identified under areas of microgyric cortex (Lyon and Beaugerie, 1988, Evrard et al., 1989). Several familial, probably genetically determined cases have been reported.
Isolated cases involving large deletions of the critical region of chromosome 22q11.2 have been described (Bingham et al., 1998, Worthington et al., 2000), with the suggestion that this may be a contributing factor in the occurrence of PMG. Other rare types of PMH have been identified in Zellweger syndrome (Raoul et al, 2003) and in other unusual syndromes such as Dellemann's syndrome (Moog et al., 2005, Pascual-Castroviejo et al., 2005).
Brain abnormalities (migration disorders) in children: clinical and radiological manifestations
E.P. Shestova, S.K. Evtushenko, E.M. Solovyova, A.V. Dushatskaya
Key words: children, brain abnormalities, migration disorders, cortical dysplasia, disorders of brain architecture.
The active introduction of modern neuroimaging examination methods (CT, MRI of the brain) into the practice of a pediatric neurologist has made it possible to significantly expand knowledge in the field of anomalies of the neuroontogenetic process and determine their role in assessing the neurological status of the child and the prognosis of the disease. The use of CT and MRI has brought the intravital study of brain structure to a higher level, switching knowledge from the formal level of “hypoxic encephalopathy” to the level of diseases caused by genetic disorders of transcription factors [2].
It has become apparent that abnormalities of brain development are the most common cause of childhood neurological disability. “Failures” of the neuroontogenetic process mostly represent a multifactorial pathology of the embryonic period. Their monogenic inheritance is observed infrequently, in no more than 1% of cases. Most of the congenital defects of the nervous system are formed under the influence of damaging agents during critical periods of embryonic development of organs and systems (according to P.G. Svetlov), and the nature and type of the defect does not depend on the nature of the damaging agent (mutant gene, chemical mutagens, ionizing radiation, viruses ), but on the age of the embryo. In this regard, it is often not possible to definitively determine the reason for the arrest of the development of an organ or system.
According to modern ideas about disorders of embryogenesis, the following factors can affect the processes of formation of the nervous system:
- exogenous toxins;
- genetic causes (sporadic cases predominate over inherited ones);
- endogenous toxins (metabolic disorders in the mother, including increased temperature of the maternal body);
- infectious pathogens (cytomegalovirus, toxoplasma, listeria, etc.).
Among the listed reasons contributing to the occurrence of developmental defects, the most influential is the exogenous factor. Currently, the fact of global pollution of the human environment with toxic chemicals is indisputable, which has led to their significant accumulation in the biosphere and further entry into the body with food, water and air. The quantitative volume of newly emerging mutations can increase under the influence of mutagenic environmental factors, especially such as ionizing radiation, active chemical compounds, and some biological factors. This creates real prerequisites and conditions for the occurrence of environmental damage to the population, manifested primarily in the embryotoxic effect. The acceleration of scientific and technological progress has fundamentally changed the human environment. Firstly, many factors appeared in it that people had not previously encountered (for example, 60 thousand new chemicals). Secondly, the environment is changing at a very fast pace. The genotype of populations does not have time to adequately respond to environmental changes. This leads to the fact that in altered environmental conditions, hereditary diseases of a new class arise - ecogenetic diseases. Their essence boils down to the fact that a certain proportion of the population has an allele that exhibits a pathological effect when exposed to a specific environmental factor. The new class of ecogenetic diseases is still largely undeciphered, although it is of great importance from the point of view of occupational medicine, food hygiene, and the environment [2]. Thus, an increase in the number of brain defects due to spontaneous mutations due to the harmful influence of ecology is increasingly gaining a place in views on the etiology of brain abnormalities.
MRI plays a leading role in the diagnosis of many brain malformations. With the help of this examination, it became possible to identify intravital brain abnormalities in a sick child that had not previously been identified and, accordingly, were not considered as a cause of neurological disorders in children.
This research method acquires particular value in the early stages of postnatal development, since during the neonatal period the disturbed condition of a child with a brain defect is unreasonably interpreted as intrauterine meningoencephalitis, hypoxic or birth damage to the nervous system.
With the help of radiological research methods, abnormalities in brain development associated with disturbances in the shape and structure (architectonics) of the gray matter are more clearly visualized. To a lesser extent, this concerns disturbances in the structure of white matter. Therefore, most of the defects are associated with an abnormality of the cerebral cortex and are collectively called cortical dysplasias.
Brain defects can develop at all stages of the embryonic and partly in the fetal period. If a “breakdown” occurs during the formation of the prosencephalon (2-3 months of intrauterine development), then gross, often “visible to the eye” defects occur, which can be judged by the presence of concomitant facial dysostosis in the patient. These, for example, include holoprosencephaly, which forms at a strictly fixed time for its development - days 22-24 of intrauterine development. Such brain anomalies can be diagnosed without additional examination methods, since they are associated with severe specific neurological symptoms, facial dysostosis, and bone disorders.
Of greatest interest to the clinician are malformations of the brain that are formed as a result of abnormal processes of brain development from the 6th to the 20th week of intrauterine development, occurring during the following periods [8]:
- 3-4th month of gestation - proliferation process (starts from the 7th week);
- 3-5th month of gestation - centrifugal migration (starts from the 8th week);
- 5th month of gestation - neural organization (lamination, gyritation and sulcation).
Developmental anomalies that form during these periods are called dismigration disorders of the brain, or anomalies (diseases) of neural migration, transmantle dysplasia, disorders of brain architectonics, etc. Often these names and concepts are mixed with each other, and sometimes it is difficult to separate them from each other, since there are no specific symptoms for each disorder.
Researchers have discovered a “gene mutation that disrupts the migration and separation of neurons during the formation of brain structures (RELN gene, chromosome 7q22). The product of this gene has been identified—the protein reelin glycoprotein, which serves as a “conductor” for neurons” [3].
Dismigration disorders are manifested by symptoms of moderate and severe organic damage to the nervous system. Their symptoms largely depend on the location of brain structure disorders and their prevalence. However, MRI images of obvious brain abnormalities are not always associated with clinical manifestations of pathological symptoms, which is currently poorly explained. Probably, such cases are exceptions to the rules, which by their existence confirm these rules. The degree of neurological disorders can be affected by the prevalence (area, volume) of disorders of brain architecture, known in morphology as malposition and malorientation. Unfortunately, these changes are poorly visualized on MRI.
In general, the symptoms of brain abnormalities are clearly expressed, but not very specific. As a rule, more severe disorders manifest themselves already in the neonatal period with adaptation disorders and convulsive syndrome. As the child grows, a delay in psychomotor development, focal neurological deficit of varying degrees of severity, and often a syndrome complex of cerebral palsy are more and more clearly formed. Epileptic seizures in children appear at different age periods, but more often they occur in the first decade; they are often resistant to therapy [5]. Often many syndromes are combined with each other, but they can also be an isolated manifestation of a malformation of the brain.
The age of onset of epileptic seizures due to abnormalities in brain development can be influenced by the so-called critical period of child development. Not everything here is clear and understandable, but it is obvious that epileptic seizures appear at different age periods with visual visualization of the lesion on MRI.
Why do epileptic seizures caused by brain developmental defects appear at different age periods? The answer to this question can be partially given by the following statement by Yu.E. Veltishcheva: “Changes in the genetic program for the regulation of normal growth and development of a child manifest themselves in discreteness, i.e. conditional isolation of individual periods of ontogenesis. ...In postnatal development, critical periods, or phases, separate individual turning points of ontogenesis - they represent a kind of “milestones” in the overall program of human development” [4]. The identification of postnatal critical periods, or phases, of development was proposed by Yu.E. Veltishchev et al. back in 1983. In doing so, the authors relied on objective and well-known manifestations of gene repression and gene switching. Such critical periods of the child include:
- neonatal;
- period 3-6 months;
- second year of life;
- age 6 years;
- puberty.
The concept of cortical dysplasia combines the following variants of neuronal organization disorders: lissencephaly (agyria), pachygyria, micropolygyria, schizencephaly and transmantle dysplasia. All of them can be focal or generalized.
One of the manifestations of abnormal neuronal and glial organization is transmantle dysplasia. According to the definition of AJ Barcovich, focal transmantle dysplasia is an area of architectural disturbance that was formed as a result of abnormal development of a stem cell and is located from the wall of the brain ventricle to the cortex [5]. In the literature, there is such a thing as generalized focal transmantle dysplasia - a large area of the brain with wide irregular gyri and sulci, with areas of hyperintensity and isointensity of gray matter to white matter. The white matter may also be abnormal. Clinically, transmantle dysplasia manifests itself as a gross focal neurological defect and epilepsy.
Lissencephaly (agyria) and pachygyria are underdevelopment of the cerebral convolutions with a smooth surface of the cerebral hemispheres, which can be total or focal. In general, lissencephaly is characterized by mental retardation and early onset of epilepsy such as infantile spasms. It occurs in both girls and boys. A specific EEG pattern is often recorded - generalized beta activity of high amplitude. Total agyria is accompanied by banded heterotopia, known as the “double cortex” syndrome.
Two morphological types of lissencephaly have been described [7].
1st - Bilschowski type, which is characterized by a 4-layer bark. The fourth layer is formed from heterotopic neurons. This type is often associated with other anomalies - heterotopia, macro- and microgyria, schizencephaly, etc. Clinically, patients have hypotension, mental retardation, epileptic paroxysms of the type of infantile spasms, myoclonus, Lennox-Gastaut syndrome. This type has genetic and chromosomal determination. It is the main morphological feature of Warburg and Seckel syndromes (dwarfism with bird-headedness), Miller-Dilker and Norman-Roberts syndromes (epilepsy, mental retardation, facial dysmorphism and other stigmas) associated with deletion of the 17th chromosome.
Type 2 - Walker's lissencephaly with complete absence of the cortical layer. Combined with hypoplasia of the cerebellum, pons, eye anomaly and other brain malformations. May occur in Dendy syndrome, Walker-Warburg syndrome.
Microgyria (micropolygyria) is a multitude of small, short, shallow convolutions [7]. Focal microgyria of varying area is more common. It may be the structural basis of many genetic and chromosomal syndromes (Dandy-Walker, Arnold-Chiari, Zellweger, neonatal adrenoleukodystrophy, etc.). Microgyria is a morphological defect in Foix-Chavany-Marie syndrome (mental retardation and pseudobulbar palsy).
Microgyria (polymicrogyria) is another variant of cortical dysplasia, indicating an area of many small, shallow convolutions with a violation of the structure of the gray matter. Polymicrogyria, which is located on both sides of the Sylvian fissure, has specific clinical manifestations and is called “congenital bilateral perisylvian syndrome” [7]. Its clinical symptoms are: congenital central diplegia of the facial, pharyngeal and masticatory muscles, 100% impairment of tongue movement in combination with mental retardation and epilepsy. Seizures usually debut in the first year of life. By their nature, they can be either focal or generalized, sometimes similar to infantile spasms, and are resistant to anticonvulsant therapy.
In the literature you can find another term - “focal cortical dysplasia” (FCD), which claims to be independent. But, according to many researchers [1, 5], this is nothing more than focal micropolygyria. FCD is a partial disruption of the neuroontogenetic processes of neuronal migration, which results in the formation of pathological cortical areas (giant neurons and bizarrely shaped astrocytes, the phenomenon of malposition and malorientation). The area of primary localization of FCD is the frontal and temporal regions of the brain.
The main nosological criteria for paroxysms:
- epileptic seizures are short-lived (no more than 1 minute);
- complex partial seizures with minimal signs of post-seizure confusion;
- secondary generalization of seizures occurs faster than with temporal lobe epilepsy;
- often demonstrative and unusual motor phenomena;
- high frequency of automatisms in the initial phase of seizures;
- frequent sudden falls.
Focal cortical dysplasia is characterized by pronounced, demonstrative and sometimes unusual motor phenomena (gesture automatisms (de novo), pedaling like marking time) accompanying seizures. During an attack, motor manifestation is pronounced, including atypical postural settings such as bilateral or unilateral tonic postures and/or atonic episodes. Between seizures, the EEG sometimes shows unusual and extremely active focal epileptic discharges in the form of repeated spikes.
A peculiar anomaly of neuronal migration is schizencephaly - a total pathology with the formation of glial migration trajectories, extending from the ventricles to the cerebral cortex. This developmental defect is clearly visualized on brain tomograms in the form of fissures of varying degrees of severity. In the classic version, these gaps in the cerebral cortex end in “open lips” (Fig. 3b). The walls of the cracks are lined with pathologically thickened bark. With this defect, cerebrospinal fluid dynamics is not impaired, it is fully compensated. Near the fissures, as a rule, there are foci of heterotopia and/or polymicrogyria [7]. Clinical manifestations of schizencephaly are associated with many neurological symptoms and syndromes such as:
- hemiplegia (with unilateral location);
- tetraparesis (with bilateral location);
- convulsive syndrome;
- severe delay in psychomotor development.
The most common variant of migration disorders is heterotopia - a cluster of neurons that have stopped in various abnormal places along the way to the cerebral cortex. This stop occurs no later than the 5th month of intrauterine development. An isolated area of nodular mass is called a “heterotopion.” The following variants of heterotopia are currently described:
- subependymal nodular (nodular) heterotopia;
- band (layered, laminar) heterotopia;
- isolated (single) heterotopia;
- "double cortex" syndrome.
Subependymal nodular (nodular) heterotopia is associated with a mutation in the FLN1 gene (Xq28). In this case, boys die, and girls are born with nodular heterotopia. Subependymal heterotopia can be single or multiple. It is most often localized in the region of the ventricular triangle and the temporal and occipital horns of the ventricles of the brain. Often visualized foci of subependymal heterotopia are regarded as focal ischemic lesions of the brain or as calcifications, which complicates the diagnosis of the disease. In patients with isolated subependymal heterotopia, seizures usually appear in the second decade. When heterotopion is localized in the subcortical region, the cerebral cortex is often abnormal, with thin and small convolutions. Such patients have varying degrees of delayed psychomotor development, depending on the size and location of the heterotopion. Paroxysms develop in almost all patients.
Most often, solitary heterotopions occur in patients. Unlike hamartomas, in tuberous sclerosis they do not accumulate contrast agent.
Ribbon (layered, laminar) heterotopia is characterized by an accumulation of heterotopions parallel to the supposed cerebral cortex. This variant of heterotopia is called the “double cortex” syndrome. It can be found in X-linked lissencephaly, which results from a mutation in the DCX gene (XLIS) Xq22.3-q23). In this case, girls are born with “double cortex” syndrome.
We present a clinical and radiological analysis of 22 children aged from 2 months to 1 year, who were in the neurological department of the regional children's clinical hospital in Donetsk from 2003 to 2005.
Patients with moderate and severe disorders of the nervous system were selected for analysis and underwent MRI of the brain in T1 and T2 modes.
All patients had moderate and severe delayed psychomotor development, lack of reduction of tonic reflexes, delayed formation of righting reflexes of varying severity, and polymorphic epileptic seizures. Seizures were represented by Ohtahara and West syndromes, generalized tonic paroxysms, focal and generalized myoclonus. In the neonatal period, 86% of patients were diagnosed with depression syndrome, 32% with agitation syndrome, and 14% with convulsive syndrome. In 65% of children, these newborn conditions were regarded only as hypoxic brain damage, and in 12% - only as a manifestation of intrauterine infection. Not a single child had a radiological examination or was diagnosed with a brain defect during the neonatal period.
The most common sign of gross disorders of the nervous system was focal transmantle malformations. On MRI and CT scans in these children, direct and indirect signs of impaired neuronal migration and various combinations of them were found. Indirect signs of brain defects included an increase in the size of the interhemispheric fissure, subarachnoid spaces, ventriculomegaly without clinical manifestations of hydrocephalus. Indirect signs accompanied almost all gross immigration violations. A pronounced expansion of the interhemispheric sulcus and subarachnoid spaces (as an indicator of brain underdevelopment) was more often found in the anterior parts of the hemispheres.
Among the direct signs of brain abnormalities in patients with severe disorders of the nervous system, the most common were clefts of the brain, which we regarded as a variant of schizencephaly. They were of different sizes, on one or both sides.
Another common direct sign of brain abnormalities was various disturbances in the pattern of convolutions (sulcations). They were noted in the form of areas of pachygyria (less often diffuse), when obvious signs of a simplified pattern of cerebral convolutions were identified. Less common was focal micropolygyria.
Another frequent manifestation of focal transmantle dysplasia in this category of patients were MR signal anomalies of varying shape and area, noted along the entire mantle of the brain. These changes are difficult to interpret as a specific developmental defect. Perhaps this is one of the options for violating the architectonics.
Along with pachygyria, ventriculomegaly, and widening of the interhemispheric sulcus, heterotopias were noted in some patients. More often we encountered subependymal heterotopia (nodular accumulation of neurons around the lateral ventricles), which was often incorrectly interpreted as a consequence of ischemic brain damage or as calcifications due to an intrauterine infection. Such patients developed convulsive syndrome at different stages of development. Heterotopia in the form of single nodules between the cortex and ventricles of the brain (subcortical heterotopia) was rarely encountered and always in combination with other transmantle disorders. Isolated heterotopia without other brain abnormalities was not encountered in the examined children.
It should be noted that all patients had different combinations of different variants of dismigration anomalies, which probably determined the severity of neurological symptoms. Experience shows that isolated developmental defects do not manifest themselves with such pronounced neurological symptoms, which are sometimes completely absent. In this case, single developmental anomalies, for example heterotopia, are defined as a finding. But the threat of paroxysms in a child at the next stage of development remains.
We believe that the following standards will help a practitioner in diagnosing brain abnormalities.
- The absence of a critical period, including severe hypoxia in the neonatal period, in the presence of pathology of the neurological status, makes it possible to assume an abnormality in brain development, especially in a full-term newborn.
- Particular attention to an increase in the size of the ventricles of the brain: in a premature baby, isolated ventriculomegaly is most often a consequence of hypoxic damage to the nervous system. In the full-term newborn, ventriculomegaly is often a radiological manifestation of a brain abnormality.
- For differential diagnosis with intrauterine encephalitis, specific immunological studies of cerebrospinal fluid will help.
- If a seizure syndrome is present in the neonatal period, the child should undergo radiological examination for the presence of brain abnormalities.
- Hypotonia during the neonatal period is a common symptom of gross malformations of the brain.
- Delayed psychomotor development and impaired development of postural reflexes are often also syndromes of brain abnormalities.
Identifying brain malformations as early as possible in life cannot be overemphasized. Without timely diagnosis of such developmental anomalies, the patient will be doomed to receive therapy for hypoxic brain damage or intrauterine infection until the diagnosis of the brain defect becomes obvious at an older age of the child.
Literature 1. Alikhanov A.A. Neuroradiological model of various variants of neuronal migration disorders // Journal of Neurology and Psychiatry. - 2004. - No. 10. — P. 81-85. 2. Bochkov N.P. The contribution of genetics to medicine // Journal of Neurology and Psychiatry. - 2002. - No. 2. — P. 3-15. 3. Veltishchev Yu.E. Current directions of scientific research in pediatrics // Russian Bulletin of Perinatology and Pediatrics. - 2003. - No. 1. — P. 5-11. 4. Veltishchev Yu.E., Yuryeva E.A. On the importance of laboratory diagnostic methods for prophylactic (preventive) pediatrics // Russian Bulletin of Perinatology and Pediatrics. - 2000. - No. 5. — P. 6-14. 5. Barkovich AJ, Kuzniecky RI, Bollen AW, Grant PE Focal transmantle dysplasia: A specific malformation of cortical development // Neurology. - 1997. - V. 49, No. 4. 6. Cohen MM, Jr. The Child With Multiple Birth Defects. — Second edition. - New York: Oxford University Press, 1997. - 267 p. 7. Gordon Neil. Epilepsy and Disorders of Neuronal Migration. I Introduction // Developmental Medicine and Child Neurology. - 1996. - V. 38. - R. 1053-1057. 8. Volpe J. Neurology of newborns. — New York: Raven Press, 1986.