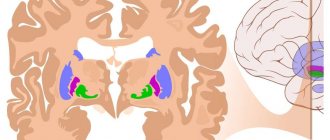
Полимикрогирия, агирия и пахигирия относятся к врожденным порокам развития головного мозга, также называются мальформациями головного мозга.
Данные аномалии возникают в результате нарушенного процесса формирования головного мозга или отдельных его структур во время пренатального периода, и представляют собой аномальные изменения в строении структур головного мозга.
Не существует точных данных о распространенности врожденных пороков головного мозга, однако наиболее распространенной считается полимикрогирия.
О каждой мальформации подробней:
- Полимикрогирия – редко встречающаяся патология развития коры головного мозга, при которой на поверхности больших полушарий формируется значительное количество мелких и недоразвитых извилин. Часто сочетается с другими генетическими патологиями и аномалиями развития коры головного мозга. Наиболее часто патология развивается в области сильвиевой борозды (60% случаев заболевания), однако может возникать в любой части коры головного мозга. Патология бывает диффузной, мультифокальной и очаговой. Также ее можно разделить на одностороннюю (40% случаев) и двустороннюю (60% случаев), симметричную и ассиметричную.
- Агирия (лиссэнцефалия) – аномалия формирования коры головного мозга, при которой извилины недоразвиты, слабо выражены, либо отсутствуют, архитектоника коры нарушена. Внешний вид головного мозга ребенка идентичен внешнему виду мозга плода на 3-4 месяце пренатального периода. Агирия является тяжелой формой лиссэнцефалии. Может быть как идиопатическим заболеванием, так и сопровождать другие патологии (синдром Миллера-Дикера, синдром Нормана-Робертса, синдром Уокера-Варбурга, врожденную мышечную дистрофию Фукуямы).
- Пахигирия является редкой аномалией развития центральной нервной системы, при которой в коре больших полушарий головного мозга формируется небольшое количество широких и плоских извилин. При пахигирии основные извилины укрупнены, а вторичные и третичные отсутствуют, борозды укорочены и выпрямлены, архитектоника церебральной коры нарушена. Данную патологию рассматривают как «неполную», более легкую форму лиссэнцефалии.
Мальформации головного мозга: полимикрогирия, агирия и пахигирия
Полимикрогирия, агирия и пахигирия относятся к врожденным порокам развития головного мозга, также называются мальформациями головного мозга. Данные аномалии возникают в результате нарушенного процесса формирования головного мозга или отдельных его структур во время пренатального периода, и представляют собой аномальные изменения в строении структур головного мозга.
Не существует точных данных о распространенности врожденных пороков головного мозга, однако наиболее распространенной считается полимикрогирия.
О каждой мальформации подробней:
Определение
В том случае, если миграция нейронов нарушена, кора перестает нормально образовывать извилины и борозды. Нарушается ее многослойная структура. Лиссэнцефалия – врожденный порок развития коры головного мозга ребенка, при котором формируется тонкая, четырехслойная кора и отсутствуют извилины и борозды. Полное отсутствие извилин называется агирией, и это наиболее тяжелая форма, которой проявляется лиссэнцефалия. В том случае, если извилины существуют, но сильно деформированы и недоразвиты, это состояние называют пахигирией. На первом фото – агирия, на следующем – сравнение извилин и борозд коры головного мозга здорового ребенка и больного лиссэнцефалией.
Лиссэнцефалия и агирия может быть самостоятельным дефектом развития коры у ребенка, или сопутствовать таким врожденным заболеваниям, как синдромы Миллера-Дикера и Нормана-Робертса, которые также являются редкими патологиями.
Провоцирующие факторы
Основная причина развития полимикрогирии, пахигирии и агирии в неправильном протекании процесса нейрональной миграции, причиной чего являются:
Полимикрогирия может быть вызвана цитомегаловирусной инфекцией, недостаточным насыщением плаценты кислородом, а мутации происходят в генах COL18A1 (21q22.3), PAX6 (11p13), K1AA1279 (10q22.1), TUBB2B (6p25), EOMES (3p21.3-p21.2), RAB3GAP1 (2q21.3), SRPX2 (Xq21.33-q23).
Агирия может возникнуть из-за вирусных инфекций, которые проникают в матку или в плод в период первого триместра, недостаточного кровоснабжения мозга плода в первые месяцы беременности. Среди генетических факторов можно назвать мутации в гене RELN (7q22), генах X хромосомы, 17 хромосомы.
При пахигирии возникают мутации в генах LIS1 (17p13.3) и DCX (Xq22.3-q23).
Диагностика заболевания
Первичная диагностика лиссэнцефалии не представляет особых затруднений. Для этого применяются те методы, которые существуют в каждом крупном городе:
- УЗИ головного мозга плода в возрасте 22-27 недель, после основного процесса формирования борозд и извилин;
- компьютерная томография, позволяющая быстро определить основные варианты нарушения топографии серого и белого вещества головного мозга;
- С помощью МРТ можно определить тип заболевания, и вывести предположительный диагноз о генетических нарушениях;
- Электроэнцефалография (ЭЭГ), с помощью которой у ребенка можно получить характерную картину электрической активности неправильно сформированной коры больших полушарий.
Характерная симптоматика
Для полимикрогирии характерны такие симптомы:
Пахигирия проявляет себя в возрасте до 2 лет. Основными ее признаками являются:
Агирия проявляется к 3-5 месяцу жизни ребенка, реже к 8-9 месяцу, основной симптом патологии выражается в стремительном отставании ребенка в физическом и умственном развитии.
Также аномалия может проявлять себя следующими признаками:
Фото мозга в разрезе и на МРТ ребенка с диагнозом агирия
Как распознать
В некоторых случаях порок развития мозга можно распознать еще внутриутробно, при проведении УЗИ плода. Во время беременности оценивать формирование борозд и извилин у плода можно не ранее 20 недели, так как на ранних сроках беременности гладкий мозг — это нормальная стадия развития плода. Также проблему можно заметить при проведении УЗИ мозга (нейросонографии) в первые месяцы жизни ребенка. При рождении на проблемы с мозгом у ребенка могут указать пороки развития пальцев, особенности строения головы и лица, порок сердца и др.
Статья по теме
Мозг себе на уме. Что за «существо» живёт в нашей черепной коробке? Но бывает и так, что при благоприятном течении беременности и родов ребенок с такой патологией расценивается как здоровый и первое время не вызывает беспокойства у родителей. Однако постепенно становится заметной задержка развития, что часто и является первой жалобой. При осмотре в большинстве случаев врач обращает внимание на уменьшенный размер головы.
В дальнейшем присоединяются эпилептические приступы, иногда родители обращаются к врачу уже в связи с их появлением. Начало эпилептических приступов отмечается, когда ребенок достигает возраста 3-5 месяцев, реже они начинаются после 9 месяцев. Первым типом приступов бывают эпилептические спазмы. Купируются такие приступы специальными препаратами. Чтобы их правильно подобрать для ребенка, следует провести целый ряд исследований: ЭЭГ, видео-ЭЭГ мониторинг со сном.
Ребенок с такой патологией дальше будет все сильнее отставать в развитии (и умственном, и физическом). Эпилептические приступы будет все сложнее останавливать и лечить. В наиболее тяжелых случаях ребенок не приобретает практически никаких навыков (не держит голову, не переворачивается, не сидит и др., отсутствует речь). Формируются парезы и параличи конечностей (как правило, с повышением мышечного тонуса). Также возможны нарушения работы внутренних органов (пороки развития сердца, почек, желудочно-кишечного тракта, половой системы, аномалии пальцев и расщелина нёба). Например, такая тяжелая картина характерна для лиссэнцефалии при синдроме Миллера-Дикера и некоторых других формах заболевания.
«Вскрытие» мозга. Допинги для интеллекта разделят людей на богов и глупцов? Подробнее
Постановка диагноза
Для диагностики мальформаций головного мозга применяют такие исследования:
При полимикрогирии кора головного мозга аномально тонкая, увеличены субарахноидальные пространства и желудочки мозга. Поражения
могут располагаться с одной или двух сторон, могут быть локальными или обширными. Кора может выглядеть аномально толстой из-за большого количества мелких извилин, которые сливаются и образуют толстые, глубокие массы.
Пахигирию определяют по результатам МРТ, на которых видны утолщения коры головного мозга, небольшие извилины, и недоразвитая сильвиевая борозда.
В пренатальный период определить мальформации мозга возможно при помощи УЗИ плода. При подозрениях на аномалии в развитии направляют на дополнительные исследования (МРТ, генетический анализ генов-кандидатов).
Профилактика
Единственное, что может каким-то образом предотвратить развитие проблемы, — это тщательное планирование беременности. Будущим родителям обязательно стоит пройти консультацию генетика, чтобы исключить риск мутаций. Также стоит сдать анализы на TORCH-инфекции.
При появлении в семье ребенка с лиссэнцефалией генетическое обследование при планировании дальнейших детей необходимо, так как с первых описаний заболевания известны случаи его выявления у нескольких детей в одной семье. В этих случаях особенно важно провести генетические исследования у больного ребенка. Какие именно, подскажет врач-генетик. После всех исследований надо будет пройти консультацию с врачом и решить вопрос о безопасном планировании потомства.
Медицина практически бессильна
Эффективные методы лечения мальформаций головного мозга отсутствуют в современной медицине. Терапия направлена на облегчение симптомов, и поддержание функционирования организма на приемлемом уровне.
При полимикрогирии и пахигирии рекомендованы такие методы облегчения состояния пациентов:
При агирии неэффективными считаются классические методы терапии: прием ноотропных препаратов, массаж, физиотерапия.
Наибольшую эффективность показывает лечение противосудорожными препаратами и миорелаксантами центрального действия, которые позволяют снизить тонус в скелетных мышцах.
Часто эпилепсия, сопровождающая патологии развития головного мозга, оказывается резистентной к терапии, и тогда рекомендуется использовать комбинацию из двух препаратов.
Проводятся исследования по лечению данных аномалия при помощи пересадки стволовых клеток, трансплантации факторов роста.
Прогноз и продолжительность жизни
Пациенты с полимикрогирией, имеющие легкие формы патологии, могут дожить до взрослого возраста. При тяжелых формах летальный исход наступает в раннем и молодом возрасте по причине пневмонии или судорог.
Для пациентов с пахигирией не существует единого прогноза. Он может варьироваться в каждом конкретном случае и зависит от тяжести патологии головного мозга, и интенсивности симптомов. Но обычно дети с пахигирией живут не дольше 20 лет.
Недоразвитие мозга у детей при агирии необратимое и прогноз неблагоприятный. Большая часть пациентов не доживает до возраста 5 лет. У всех больных присутствует полная инвалидизация.
Из-за угнетенных глотательных рефлексов часто требуется зондовое питание. Летальный исход может наступить по причине заболеваний, возникающих на фоне основной патологии: парез кишечника, сердечно-сосудистая недостаточность, дыхательная недостаточность, атрофия дыхательной мускулатуры, гипостатическая пневмония.
Источник
Пахигирия головного мозга симптомы
Термины микрогирия
и
полимикрогирия
(ПМГ) описывают патологические изменения коры головного мозга, представленные множеством коротких, неглубоких извилин, формирующих патологический рисунок, который макроскопически может различаться. В отдельных случаях при оценке поверхности мозга широкие извилины трудно отличить от участков пахигирии, однако кортикальная лента имеет типичный полимикрогирический вид со слиянием молекулярных слоев (Friede, 1989, Harding и Сорр, 1997). Микрогирия может быть генерализованной, односторонней, многоочаговой и очаговой.
Различают два основных гистологических типа полимикрогирии (ПМГ), но часто встречаются переходные или смешанные формы. Наиболее распространенным безусловным нарушением развития является микрогирия с неразделенными слоями (Ferrer, 1984, Harding и Сорр, 1997), при которой между белым веществом и молекулярным слоем имеется единственный волнистый клеточный слой.
При классической четырехслойной микрогирии первый слой из малых пирамидальных клеток, расположенный ниже молекулярного слоя, отделен низкоклеточным слоем от глубокого, содержащего большие пирамидальные клетки. Низкоклеточный слой соответствует IV и V нормальным слоям, с которыми он сливается у границ микрогирической коры (Williams и Caviness, 1984, Evrard et al., 1989). Указанное явление позволяет высказать предположение о возникающих на пятом или шестом месяце беременности постмиграционных нарушениях с вторичными ламинарными некрозами, развивающимися в результате недостаточности кровообращения и гипоксии. Данная интерпертация подкрепляется тем, что классическая ПМГ развивается вследствие патологических событий, встречающихся после середины текущей беременности, таких как тяжелая травма, интоксикация оксидом углерода или материнская асфиксия (Larroche, 1986, Barth, 2003b), и только у одного из монозиготных близнецов (Sugama и Kusano, 1994). Evrard et al. (1989) выделили два пиковых периода для недостаточности кровообращения: между 20 и 24 неделями и в течение последних 10 недель гестации.
Недостаточность кровотока также может отвечать за микрогирию при фетальных инфекциях (Marques-Dias et al., 1984), способных вызвать повреждение сосудов (Barkovich и Lindan, 1994) или циркуляторную недостаточность, а также за порэнцефалические дефекты. Однако отдельные ПМГ могут формироваться незадолго до конца процессов миграции и возникать в результате гибели поверхностных слоев, повреждая глиальные проводники до того, как поздно мигрирующие клетки закончат процесс перемещения (Dvorak et al., 1978). В этих случаях нейроны, остановленные в процессе миграции, определяются под участками микрогирической коры (Lyon и Beaugerie, 1988, Evrard et al., 1989). Зарегистрировано несколько семейных, вероятно, генетически детерминированных случаев.
Были описаны отдельные случаи, связанные с большими делециями критической области хромосомы 22q11.2 (Bingham et al., 1998, Worthington et al., 2000), с предположением, что это может быть фактором, способствующим возникновению ПМГ. Другие редкие типы ПМГ выявлялись при синдроме Цельвегера (Raoul et al, 2003) и при других необычных синдромах, таких как синдром Деллеманна (Moog et al., 2005, Pascual-Castroviejo et al., 2005).
Аномалии головного мозга (миграционные нарушения) у детей: клинико-радиологические проявления
Е.П. Шестова, С.К. Евтушенко, Е.М. Соловьева, А.В. Душацкая
Ключевые слова: дети, аномалии головного мозга, миграционные нарушения, кортикальные дисплазии, нарушения архитектоники мозга.
Активное внедрение в практику детского невролога современных нейровизуализирующих методов обследования (КТ, МРТ головного мозга) позволило значительно расширить знания в области аномалий нейроонтогенетического процесса, определить их роль при оценке неврологического статуса ребенка и прогноза заболевания. Использование КТ и МРТ перевело прижизненное изучение структуры мозга на более высокую ступень, переключив познание с формального уровня «гипоксическая энцефалопатия» на уровень болезней, вызванных генетическим нарушением транскрипционных факторов [2].
Стало очевидным, что аномалии развития головного мозга являются наиболее частой причиной детской неврологической инвалидности. «Поломки» нейроонтогенетического процесса в большинстве своем представляют мультифакториальную патологию эмбрионального периода. Их моногенное наследование наблюдается нечасто, не более чем в 1% случаев. Большая часть врожденных пороков нервной системы формируется под воздействием повреждающих агентов в критические периоды эмбрионального развития органов и систем (по П.Г. Светлову), причем характер, вид порока зависит не от природы повреждающего агента (мутантный ген, химические мутагены, ионизирующая радиация, вирусы), а от возраста эмбриона. В связи с этим окончательно выяснить причину остановки развития органа или системы часто не представляется возможным.
Согласно современным представлениям о нарушении эмбриогенеза, на процессы формирования нервной системы могут повлиять следующие факторы:
- экзогенные токсины;
- генетические причины (преобладают спорадические случаи над наследуемыми);
- эндогенные токсины (метаболические нарушения у матери, в т.ч. повышение температуры материнского организма);
- инфекционные возбудители (цитомегаловирус, токсоплазма, листерии и др.).
Среди перечисленных причин, способствующих возникновению пороков развития, наиболее влиятельным является экзогенный фактор. В настоящее время бесспорным является факт глобального загрязнения среды обитания человека токсическими химическими веществами, что привело к значительному накоплению их в биосфере и дальнейшему поступлению в организм с продуктами питания, водой и воздухом. Количественный объем вновь возникающих мутаций может увеличиваться под влиянием мутагенных факторов среды, особенно таких, как ионизирующая радиация, активные химические соединения, некоторые биологические факторы. Это создает реальные предпосылки и условия для возникновения экологических поражений населения, проявляющихся прежде всего в эмбриотоксическом эффекте. Ускорение научно-технического прогресса принципиально изменило среду обитания человека. Во-первых, в ней появилось много факторов, с которыми человек ранее не сталкивался (например, 60 тыс. новых химических веществ). Во-вторых, среда изменяется в очень быстром темпе. Генотип популяций не успевает адекватно реагировать на изменение среды. Это приводит к тому, что в измененных экологических условиях возникают наследственные болезни нового класса — экогенетические болезни. Суть их сводится к тому, что определенная доля популяции имеет аллель, который проявляет патологическое действие при воздействии конкретного фактора среды. Новый класс экогенетических болезней до сих пор во многом еще не расшифрован, хотя он имеет огромное значение с точки зрения медицины труда, гигиены питания, окружающей среды [2]. Таким образом, увеличение числа пороков головного мозга вследствие спонтанных мутаций за счет пагубного влияния экологии все больше завоевывает место во взглядах на этиологию аномалий головного мозга.
Ведущая роль в диагностике многих пороков развития головного мозга принадлежит МРТ. С помощью этого обследования появилась возможность прижизненно выявить у больного ребенка такие аномалии мозга, которые ранее не определялись и соответственно не рассматривались в качестве причины неврологических расстройств у детей.
Особую ценность этот метод исследования приобретает в ранние сроки постнатального развития, так как в период новорожденности нарушенное состояние ребенка с пороком головного мозга необоснованно трактуется как внутриутробный менингоэнцефалит, гипоксическое или родовое повреждение нервной системы.
С помощью радиологических методов исследования четче визуализируются аномалии развития головного мозга, связанные с нарушением формы и строения (архитектоники) серого вещества. В меньшей степени это касается нарушения строения белого вещества. Поэтому большую часть пороков связывают с аномалией коры головного мозга и называют обобщенно корковыми дисплазиями.
Пороки головного мозга могут развиться на всех этапах эмбрионального и частично в фетальном периоде. Если «поломка» происходит во время формирования прозэнцефалона (2-3-й месяц внутриутробного развития), то возникают грубые, нередко «видимые глазом» пороки, о которых можно судить по наличию у больного сопутствующего лицевого дизостоза. К ним, например, относится голопрозэнцефалия, формирующаяся в строго фиксированные для ее развития сроки — 22-24-й дни внутриутробного развития. Такие аномалии головного мозга можно диагностировать и без дополнительных методов обследования, так как они сопряжены с грубой специфической неврологической симптоматикой, лицевым дизостозом, костными нарушениями.
Наибольший интерес для клинициста представляют пороки развития головного мозга, формирующиеся вследствие аномальных процессов развития мозга с 6-й по 20-ю неделю внутриутробного развития, происходящих в следующие периоды [8]:
- 3-4-й месяц гестации — процесс пролиферации (начало с 7-й недели);
- 3-5-й месяц гестации — центробежная миграция (начало с 8-й недели);
- 5-й месяц гестации — нейронная организация (ламинация, гиритация и сулькация).
Аномалии развития, формирующиеся в эти периоды, получили названия дисмиграционных нарушений головного мозга, или аномалий (болезней) нейронной миграции, трансмантийных дисплазий, нарушений архитектоники головного мозга и т.д. Часто эти названия и понятия смешиваются друг с другом, и порой их трудно разделить между собой, так как для каждого нарушения отсутствует специфическая симптоматика.
Исследователями обнаружена «мутация гена, нарушающая миграцию и расслоение нейронов при закладке структур мозга (RELN-ген, хромосома 7q22). Идентифицирован продукт этого гена — белок реелин-гликопротеид, служащий «проводником» для нейронов» [3].
Дисмиграционные нарушения проявляются симптомами средней и тяжелой степени органического поражения нервной системы. Их симптоматика во многом зависит от локализации нарушений структуры мозга и от их распространенности. Тем не менее, картинки на МРТ с явной аномалией головного мозга не всегда связаны с клиническими проявлениями патологических симптомов, что в настоящее время малообъяснимо. Вероятно, такие случаи являются исключением из правил, которые своим существованием и подтверждают эти правила. На степень неврологических расстройств может повлиять распространенность (площадь, объем) нарушений архитектоники головного мозга, известных в морфологии как мальпозиция и мальориентация. К сожалению, эти изменения плохо визуализируются на МРТ.
В целом симптоматика аномалий головного мозга четко выражена, но малоспецифична. Как правило, более грубые расстройства проявляются уже в период новорожденности нарушением адаптации, судорожным синдромом. По мере роста ребенка все более четко формируется задержка психомоторного развития, очаговый неврологический дефицит разной степени выраженности, часто — синдромокомплекс церебрального паралича. Эпилептические приступы у ребенка появляются в разные возрастные периоды, но чаще это происходит в первое десятилетие, нередко они резистентны к терапии [5]. Часто многие синдромы сочетаются друг с другом, но могут быть и изолированным проявлением порока развития головного мозга.
На возраст появления эпилептических приступов вследствие аномалий развития головного мозга может повлиять так называемый критический период развития ребенка. Не все тут ясно и понятно, но то, что эпиприступы появляются в различные возрастные периоды с наглядной визуализацией очага на МРТ, очевидно.
Почему эпиприступы, обусловленные пороками развития головного мозга, появляются в различные возрастные периоды? Ответ на этот вопрос частично может дать следующее утверждение Ю.Е. Вельтищева: «Изменения в генетической программе регуляции нормального роста и развития ребенка проявляются дискретностью, т.е. условной обособленностью отдельных периодов онтогенеза. …В постнатальном развитии критические периоды, или фазы, разделяют отдельные переломные этапы онтогенеза — они представляют собой своеобразные «верстовые столбы» в общей программе развития человека» [4]. Выделение постнатальных критических периодов, или фаз, развития было предложено Ю.Е. Вельтищевым и соавт. еще в 1983 г. При этом авторы основывались на объективных и хорошо известных проявлениях репрессии генов и генного переключения. К таким критическим периодам ребенка относятся:
- неонатальный;
- период 3-6 мес.;
- второй год жизни;
- возраст 6 лет;
- пубертатный период.
Понятие кортикальной дисплазии объединяет следующие варианты нарушения нейронной организации: лиссэнцефалия (агирия), пахигирия, микрополигирия, шизэнцефалия и трансмантийная дисплазия. Все они могут носить очаговый и генерализованный характер.
Одним из проявлений аномальной нейрональной и глиальной организации является трансмантийная дисплазия. По определению A.J. Barcovich, фокальная трансмантийная дисплазия (focal transmantle dysplasia) — это участок нарушения архитектоники, который образовался вследствие аномального развития стволовой клетки и расположен от стенки желудочка мозга до кортекса [5]. В литературе встречается такое понятие, как генерализованная фокальная трансмантийная дисплазия — большая область мозга с широкими нерегулярными извилинами и бороздами, с областями гиперинтенсивности и изоинтенсивности серого вещества к белому. Белое вещество, возможно, также аномально. Клинически трансмантийная дисплазия проявляется грубым очаговым неврологическим дефектом и эпилепсией.
Лиссэнцефалия (агирия) и пахигирия — недоразвитие мозговых извилин с гладкой поверхностью мозговых гемисфер, может быть тотальной и очаговой. В целом для лиссэнцефалии характерна умственная отсталость, раннее начало эпилепсии по типу инфантильных спазмов. Встречается как у девочек, так и у мальчиков. Часто регистрируется специфический паттерн ЭЭГ- генерализованная бета-активность высокой амплитуды. Тотальная агирия сопровождается ленточной гетеротопией, известной как синдром «двойной коры».
Описаны два морфологических типа лиссэнцефалии [7].
1-й — тип Bilschowski, для которого характерна 4-слойная кора. Четвертый слой сформирован из гетеротопических нейронов. Этот тип часто ассоциируется с другими аномалиями — гетеротопиями, макро- и микрогириями, шизэнцефалией и др. Клинически у больных отмечается гипотония, умственная отсталость, эпилептические пароксизмы по типу инфантильных спазмов, миоклоний, синдрома Леннокса — Гасто. Данный тип имеет генетическую и хромосомную детерминированность. Он является основным морфологическим признаком синдромов Варбурга и Секкеля (карликовость с птицеголовостью), синдромов Miller — Dilker и Norman — Roberts (эпилепсия, умственная отсталость, лицевой дисморфизм и другие стигмы), связанные с делецией 17-й хромосомы.
2-й тип — Walker’s лиссэнцефалия с полным отсутствием кортикального слоя. Сочетается с гипоплазией мозжечка, моста, аномалией глаз и другими мальформациями мозга. Может встречаться при синдроме Dendy, синдроме Walker — Warburg.
Микрогирия (микрополигирия) — множество мелких, коротких, неглубоких извилин [7]. Чаще встречается фокальная микрогирия различной площади. Она может являться структурной основой многих генетических и хромосомных синдромов (Денди — Уокера, Арнольда -Чиари, Цельвегера, неонатальной адренолейкодистрофии и др.). Микрогирия является морфологическим дефектом при синдроме Foix — Chavany — Marie (умственная отсталость и псевдобульбарный паралич).
Микрогирия (полимикрогирия) — еще один вариант корковой дисплазии, обозначающий участок множества мелких, неглубоких извилин с нарушением строения серого вещества. Полимикрогирия, которая располагается с обеих сторон сильвиевой борозды, имеет специфические клинические проявления и получила название «врожденный двусторонний перисильвиев синдром» [7]. Клиническими симптомами ее являются: врожденная центральная диплегия лицевой, глоточной и жевательной мускулатуры, 100%-ное нарушение движения языка в сочетании с умственной отсталостью и эпилепсией. Судороги дебютируют, как правило, на первом году жизни. По своему характеру они могут быть как фокальными, так и генерализованными, иногда по типу инфантильных спазмов, резистентны к противосудорожной терапии.
В литературе можно встретить еще один термин — «фокальная корковая дисплазия» (ФКД), претендующий на самостоятельность. Но, по мнению многих исследователей [1, 5], это есть не что иное, как фокальная микрополигирия. ФКД — частичное нарушение нейроонтогенетических процессов нейронной миграции, результатом чего является образование патологических корковых участков (гигантские нейроны и причудливой формы астроциты, явления мальпозиции и мальориентации). Область преимущественной локализации ФКД — лобные и височные отделы мозга.
Основные нозологические критерии пароксизмов:
- эпиприпадки кратковременны (не более 1 мин);
- сложные парциальные припадки с минимальными явлениями постприступной спутанности;
- вторичная генерализация припадков происходит быстрее, чем при височной эпилепсии;
- часто демонстративные и необычные двигательные феномены;
- высокая частота автоматизмов в начальной фазе припадков;
- частые внезапные падения.
Для фокальной корковой дисплазии характерны выраженные, демонстративные и порой необычные двигательные феномены (жестовые автоматизмы (de novo), педалирование по типу топтания на месте), сопровождающие припадки. Во время приступа выражена моторная манифестация, включающая атипичные позные установки по типу билатеральных либо унилатеральных тонических поз и/или атонические эпизоды. Между приступами на ЭЭГ иногда отмечаются необычные и черезвычайно активные фокальные эпилептические разряды в виде повторяющихся спайков.
Своеобразной аномалией нейронной миграции является шизэнцефалия — тотальная патология с формированием глиальных миграционных траекторий, простирающаяся от желудочков до коры головного мозга. Данный порок развития хорошо визуализируется на томограммах головного мозга в виде различной степени выраженности щелей. В классическом варианте эти щели в коре головного мозга заканчиваются «открытыми губами» (рис. 3б). Стенки щелей выстланы патологически утолщенной корой. При данном пороке ликвородинамика не нарушена, она полностью компенсирована. Возле щелей, как правило, находятся очаги гетеротопии и/или полимикрогирии [7]. Клинические проявления шизэнцефалии ассоциируются со многими неврологическими симптомами и синдромами в виде:
- гемиплегии (при унилатеральном расположении);
- тетрапареза (при билатеральном расположении);
- судорожного синдрома;
- грубой задержки психомоторного развития.
Наиболее частым вариантом миграционных нарушений является гетеротопия — скопление нейронов, остановившихся в различных аномальных местах на пути следования к коре головного мозга. Такая остановка происходит не позже 5-го месяца внутриутробного развития. Изолированный участок узловатой массы называется «гетеротопион». В настоящее время описаны следующие варианты гетеротопии:
- субэпендимальная нодулярная (узелковая) гетеротопия;
- ленточная (слоистая, ламинарная) гетеротопия;
- изолированная (одиночная) гетеротопия;
- синдром «двойной коры».
Субэпендимальная узелковая (нодулярная) гетеротопия связана с мутацией гена FLN1 (Хq28). При этом мальчики погибают, а девочки рождаются с нодулярной гетеротопией. Субэпендимальная гетеротопия может быть одиночной и множественной. Локализуется чаще в области вентрикулярного треугольника и височных и затылочных рогах желудочков головного мозга. Нередко визуализируемые очаги субэпендимальной гетеротопии расценивают как очаговые ишемические поражения мозга или как кальцинаты, что затрудняет диагностику заболевания. У больных с изолированной субэпендимальной гетеротопией судороги обычно появляются во втором десятилетии. При локализации гетеротопиона в субкортикальной области кора головного мозга часто аномальная, с тонкими и мелкими извилинами. У таких больных отмечается различная степень задержки психомоторного развития, зависящаяся от размера и местоположения гетеротопиона. Пароксизмы развиваются почти у всех больных.
Наиболее часто у больных встречаются одиночные гетеротопионы. В отличие от гамартомы, при туберозном склерозе они не накапливают контрастное вещество.
Для ленточной (слоистой, ламинарной) гетеротопии характерно скопление гетеротопионов параллельно предполагаемой коре головного мозга. Данный вариант гетеротопии получил название синдрома «двойной коры». Его можно обнаружить при X-сцепленной лиссэнцефалии, развивающейся в результате мутации гена DCX (XLIS) Xq22.3-q23). При этом девочки рождаются с синдромом «двойной коры».
Представляем клинико-радиологический анализ 22 детей в возрасте от 2 месяцев до 1 года, находившихся в неврологическом отделении областной детской клинической больницы г. Донецка с 2003 по 2005 год.
Для анализа были отобраны больные со среднетяжелыми и тяжелыми расстройствами нервной системы, которым было проведено МРТ головного мозга в режимах Т1 и Т2.
У всех больных отмечали задержку психомоторного развития средней и тяжелой степени, отсутствие редукции тонических рефлексов, задержку формирования рефлексов выпрямления различной степени выраженности, полиморфные эпилептические приступы. Судороги были представлены синдромами Ohtahara и West, генерализованными тоническими пароксизмами, очаговыми и генерализованными миоклониями. В неонатальном периоде у 86% больных был диагностирован синдром угнетения, у 32% — синдром возбуждения, у 14% — судорожный синдром. У 65% детей эти состояния новорожденных были расценены только как гипоксическое поражение головного мозга, а у 12% — только как проявление внутриутробной инфекции. Ни у одного ребенка в период новорожденности не было проведено радиологическое исследование и не был диагностирован порок головного мозга.
Наиболее частым признаком грубых расстройств нервной системы были фокальные трансмантийные пороки развития. На МРТ и КТ у этих детей находили прямые и косвенные признаки нарушения миграции нейронов, различное их сочетание. К косвенным признакам пороков головного мозга относили увеличение в размерах межполушарной борозды, субарахноидальных пространств, вентрикуломегалию без клинических проявлений гидроцефалии. Косвенные признаки сопровождали практически все грубые дисмиграционные нарушения. Выраженное расширение межполушарной борозды и субарахноидальных пространств (как показатель недоразвития мозга) чаще обнаруживали в передних отделах полушарий.
Среди прямых признаков аномалий мозга у больных с тяжелыми расстройствами нервной системы наиболее часто встречались расщелины мозга, расцененные нами как вариант шизэнцефалии. Они были различных размеров, с одной или с обеих сторон.
Еще одним частым прямым признаком аномалий головного мозга были различные нарушения рисунка извилин (сулькации). Их отмечали в виде участков пахигирии (реже диффузной), когда были определены явные признаки упрощенного рисунка мозговых извилин. Реже встречали фокальную микрополигирию.
Еще одним частым проявлением фокальной трансмантийной дисплазии у данной категории больных были различные по форме и площади аномалии МР-сигнала, отмечавшиеся вдоль всей мантии головного мозга. Эти изменения трудно трактовать как какой-то определенный порок развития. Возможно, это один из вариантов нарушения архитектоники.
Наряду с пахигирией, вентрикуломегалией, расширением межполушарной борозды у части больных отмечали гетеротопии. Чаще встречали субэпендимальную гетеротопию (узловое скопление нейронов вокруг боковых желудочков), нередко неправильно трактовавшуюся как последствие ишемического поражения головного мозга или как кальцинаты вследствие перенесенной внутриутробной инфекции. У таких больных на разных этапах развития появлялся судорожный синдром. Гетеротопию в виде единичных узелков между корой и желудочками мозга (подкорковая гетеротопия) встречали редко и всегда в сочетании с другими трансмантийными расстройствами. Изолированную гетеротопию без других аномалий мозга у обследованных детей не встречали.
Надо отметить, что у всех больных выявляли различное сочетание различных вариантов дисмиграционных аномалий, что, вероятно, и определяло тяжесть неврологической симптоматики. Опыт показывает, что единичные пороки развития проявляются не такой выраженной неврологической симптоматикой, которая иногда и вовсе отсутствует. При этом единичные аномалии развития, например гетеротопии, определяются как находка. Но угроза появления пароксизмов у ребенка на следующем этапе развития остается.
Полагаем, что в диагностике аномалий головного мозга практическому врачу помогут следующие стандарты.
- Отсутствие критического периода, в том числе тяжелой гипоксии в неонатальном периоде, при наличии патологии неврологического статуса дает возможность предполагать аномалию развития головного мозга, особенно у доношенного новорожденного.
- Особое внимание к увеличению размеров желудочков мозга: у недоношенного ребенка изолированная вентрикуломегалия чаще всего является следствием гипоксического поражения нервной системы. У доношенного новорожденного вентрикуломегалия нередко является радиологическим проявлением аномалии мозга.
- Для дифференциальной диагностики с внутриутробным энцефалитом помогут специфические иммунологические исследования ликвора.
- При наличии судорожного синдрома в неонатальном периоде ребенок должен пройти радиологическое обследование на наличие аномалий головного мозга.
- Гипотония в период новорожденности является частым симптомом грубых пороков развития головного мозга.
- Задержка темпов психомоторного развития и нарушение развития постуральных рефлексов часто также являются синдромами аномалий головного мозга.
Выявление пороков развития головного мозга в как можно более ранние сроки жизни не может быть переоценено. Без своевременной диагностики таких аномалий развития пациент будет обречен получать терапию по поводу гипоксического поражения мозга или внутриутробной инфекции до тех пор, пока диагноз порока мозга не станет очевидным в более старшем возрасте ребенка.
Литература 1. Алиханов А.А. Нейрорадиологическая модель различных вариантов нарушения нейронной миграции // Журнал неврологии и психиатрии. — 2004. — №10. — С. 81-85. 2. Бочков Н.П. Вклад генетики в медицину // Журнал неврологии и психиатрии. — 2002. — №2. — С. 3-15. 3. Вельтищев Ю.Е. Актуальные направления научных исследований в педиатрии // Российский вестник перинатологии и педиатрии. — 2003. — №1. — С. 5-11. 4. Вельтищев Ю.Е., Юрьева Э.А. О значении методов лабораторной диагностики для профилактической (превентивной) педиатрии // Российский вестник перинатологии и педиатрии. — 2000. — №5. — С. 6-14. 5. Barkovich A.J., Kuzniecky R.I., Bollen A.W., Grant P.E. Focal transmantle dysplasia: A specific malformation of cortical development // Neurology. — 1997. — V. 49, №4. 6. Cohen M.M., Jr. The Child With Multiple Birth Defects. — Second edition. — New York: Oxford University Press, 1997. — 267 p. 7. Gordon Neil. Epilepsy and Disorders of Neuronal Migration. I Introduction // Developmental Medicine and Child Neurology. — 1996. — V. 38. — Р. 1053-1057. 8. Volpe J. Neurology of newborne. — New York: Raven Press, 1986.