Causes
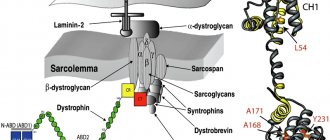
The disease is hereditary, linked to the X chromosome, so boys are almost always affected. Girls are carriers of a pathological gene (boys rarely survive to adulthood, and, moreover, are usually sterile). A change in the structure of the gene responsible for the synthesis of the dystrophin protein occurs on the chromosome.
Although the content of dystrophin in skeletal muscles is extremely small (thousandths of a percent), without it necrosis of muscle tissue quickly develops and progressive muscle dystrophy develops. If the gene is damaged in a site that completely destroys the synthesis of the dystrophin protein, Duchenne dystrophy develops. When insignificant parts of the protein are involved in the process, the disease takes the form of Becker's dystrophy.
Muscular dystrophies bilateral
In the initial period, myodystrophy on one side may predominate, but as the disease develops, the degree of damage becomes the same in the patient’s symmetrical muscles. As the disease progresses, muscle strength decreases in almost all muscles. Areas of hypertrophic muscles appear on the body of a patient suffering from muscular dystrophy. This is pseudohypertrophy, which is not associated with an increase in muscle fibers. Pseudohypertrophy of muscles is associated with swelling in the muscles of the legs or arms. Such muscles are dense, but weak.
Symptoms
The onset of symptoms begins in early childhood, usually between 1 and 3 years. Initially, there is a delay in motor development, the child begins to walk late, often stumbles when walking, and gets tired quickly. Later, constant pathological muscle fatigue develops. The child practically cannot climb stairs. The gait begins to resemble a “duck” gait.
A characteristic symptom is the “ladder” symptom: an attempt to get up from a sitting position occurs using the hands, gradually, slowly, in several stages.
Gradually, muscle atrophy begins to be noted, first in the proximal parts of the lower, then upper extremities. Later, the muscles of the pelvic girdle, hips, back, and shoulder girdle atrophy. A “wasp” waist, curvature of the spine, and protruding shoulder blades (pterygoid blades) almost always develop.
Almost always there is a characteristic symptom of progressive Duchenne muscular dystrophy - pseudohypertrophy of the leg muscles. The muscles, although increased in volume, do not have sufficient strength and are very painful to the touch.
Three stages of the disease can be distinguished: - Stage I - weakness manifests itself only with significant physical activity (usually the first year of the disease). — Stage II – difficulty climbing stairs, weakness when walking quickly develops. — Stage III – represents paralysis, muscle contractures with the inability to move independently.
According to the type of course, it is divided into: Rapid progression. The ability to move is lost quickly, within the first 4-5 years from the onset of the disease. Average rate of progression: the patient cannot move after 10 years. Slow progression: there are no pronounced motor disorders 10 years after the onset of the disease. Typically, this option is characteristic of other types of muscular dystrophies than Duchenne dystrophy.
Treatment of spinal muscular atrophy
Patients are prescribed complex conservative therapy aimed at improving the ability of nerve impulses to pass to the muscles and the functioning of the brain. For these purposes, it is recommended to take:
- nootropics;
- preparations of α-lipoic acid, acetyl-L-carnitine, α-glycerophosphocholine;
- vitamin complexes, including, first of all, B vitamins;
- means that improve metabolism.
Today, specific drugs are being developed that can affect the cause of SMA development – deficiency of a number of proteins. But at the moment they are at the testing stage. So far, the only way to at least partially provide the body with the necessary proteins is to follow a special diet. It involves eating foods rich in amino acids, namely grains, nuts, dairy products, fish, and meat. It is very useful to include spinach, broccoli, and grapefruit in the menu. Dishes made from brown rice and oats are especially valuable.
To maintain muscle tone, the following are recommended:
- exercise therapy classes;
- massage;
- physiotherapeutic treatment;
- neuromuscular stimulation.
Diagnostics
The clinical picture is very bright. Often the disease is diagnosed after clarifying a genetic history (presence of cases in the family) and a neurological examination. In the neurological status, the disappearance of knee reflexes is noted, a little later reflexes from the biceps and triceps disappear. Achilles reflexes are preserved for a long time.
Externally, deformation of the joints of the foot may be revealed, there are signs of cardiomyopathy: irregular pulse, dullness of heart sounds, dilation of the heart cavities according to EchoCG, changes in the electrocardiogram.
An important factor is the increase in the biochemical parameters of creatine phosphokinase (an enzyme indicator of muscle breakdown). The activity of this enzyme increases tens of times. There is a direct correlation between the degree of increase in enzyme activity and the severity of manifestations of Duchenne dystrophy. [!] In difficult diagnostic situations, a cytological examination is performed.
Pathological picture of the disease
Let's see what happens inside muscle cells in patients suffering from Duchenne muscular dystrophy. To do this, we will make an incision in the skin, expand it with an expander and take a small piece of muscle fiber.
In the photo: muscle tissue biopsy
In the photo: muscle tissue biopsy
A typical sign of muscular dystrophy is primarily a different diameter of muscle fibers. In a healthy person, the diameter of the muscle fibers is the same.
Characteristic signs of muscular dystrophy are atrophied and hypertrophied fibers, multiple internal nuclei and edema.
Examining stained sections of skeletal muscle, I observed denervation of myofibers, significant variation in myofibril size, and significant edema.
Explanation for the first photo:
- The pale purple color is muscle fibers in cross-section.
- Light spots both inside and outside the fibers are swelling.
- Dark dots are nuclei that the edema has shifted to the periphery.
The second photo shows normal muscle fiber from a healthy person.
The severity of muscular dystrophy according to electron microscopy is based on the following indicators:
- with a mild degree, the difference in the size of muscle fibers is moderate, the initial signs of edema (white color).
In the photo : muscle fiber biopsy for mild (A), moderate (B) and severe dystrophy (C).
- moderate severity corresponds to the movement of nuclei to the center of muscle fibers, expansion of the interfibrillar space due to increased edema between the cells.
In the photo: muscle fibers with progressive muscular dystrophy of moderate severity:
a) light purple muscle fibers;
b) light spots inside the muscle fibers - swelling, which has pushed the nuclei from the center of the cell to the periphery;
c) dark dots – muscle cell nuclei;
d) the arrow shows a muscle cell that cannot move due to a decrease in metabolic processes - it darkens towards purple.
- severe degree is characterized by extensive foci of destruction of myofibrils, their fragmentation and disorganization, the appearance of a hyaline-like substance and edema between muscle cells. Functionally, such tissue has weak strength, fatigue sets in quickly and signs of muscle fatigue develop. The photo will be presented a little lower.
This is the state of Emine’s muscles before contacting me:
Explanation for the photo “severe muscular dystrophy”:
- Muscle fibers in section are colored blue.
- The red dots are the nuclei of muscle cells.
- Edema is uncolored white.
Treatment and life prognosis
Treatment is symptomatic. Hormonal drugs are used to stop the destruction of muscle fiber, phospholipids to protect muscle cells from destruction, and elements of therapeutic exercises. Various orthopedic devices are being introduced into practice to facilitate movement. Massage is strictly contraindicated in most cases, as it can lead to accelerated muscle breakdown. Treatment of hereditary diseases is a matter of the future.
The life prognosis for patients is unfavorable. The course of the disease is progressive. Death is inevitable. As a rule, by the age of seven, severe symptoms develop, leading to complete immobility by the age of 13-14. Patients rarely live to 18-20 years of age.
What is spinal muscular atrophy and its types
This term combines several different types of hereditary diseases accompanied by limited motor abilities. This explains the fact that in some cases, disorders are detected not in infancy, but in adolescents or already mature people.
The disease was first described in 1891 by G. Werdnig and in 1892 it was identified as a separate nosological unit by J. Hoffman, thanks to whose efforts it received its second name. About half a century later, E. Kugelberg and L. Welander discovered another similar disease, developing at a later age and characterized by a more favorable course.
The following forms of pathology are distinguished:
- SMA 0;
- SMA 1 (severe form);
- SMA 2 (intermediate form);
- SMA 3 (mild form);
- SMA 4 (late form).
Cause of spinal muscular atrophy in children
What they all have in common is that the cause of their occurrence lies in the mutation of the recessive gene of chromosome 5 SMN. This leads to disruptions in the production of proteins in the body, which are the building blocks of all cells. As a result, the motor neurons of the spinal cord suffer and are gradually destroyed. Since without them it is impossible to transmit nerve impulses to muscle fibers, they gradually atrophy, which causes loss of the ability to move.
Fortunately, even if both parents have the SMN gene mutation, they have a 75% chance of having a healthy child. But almost always he will also be a carrier of this gene. Therefore, when planning a pregnancy, it is worth undergoing genetic testing, especially if there are cases of SMA in the family.
SMA 0
This is a congenital disease, the signs of which are usually detected in the maternity hospital. It is rare and is often combined with SMA-1. This species is characterized by an absolute lack of mobility, muscle weakness, lack of tendon reflexes and limited functionality of the knee joints. From the first days of life, the child suffers from breathing problems.
It is important to differentiate spinal muscular atrophy from perinatal encephalopathy and birth injuries, but if with them the condition of children gradually improves, then with SMA it does not change. Moreover, complications are often added, which almost always lead to the death of infants during the first month of life.
SMA 1 or Werding-Hoffman disease
This type of spinal muscular atrophy is characterized by a very severe course. It is usually detected before 6 months of age and is accompanied by muscle weakness and periodic spasms, which is difficult to notice due to the anatomy of children in the first year of life (the presence of pronounced subcutaneous fat).
The disease also manifests itself regularly as tremors running through the tongue, decreased gag, sucking, and swallowing reflexes. This leads to serious feeding difficulties. There is a violation of salivation and cough. The child often screams loudly.
Since the chest muscles are not developed enough, you may notice that the shape of the chest is flatter. In addition, children with this pathology lie and sleep in the “frog” position: with their shoulders and hips pulled to the sides and their limbs bent at the knees and elbows. They are able to learn to hold their head up by six months (it is often smaller than that of healthy children), but they are not able to sit independently or take an upright body position.
This form of spinal muscular atrophy may be accompanied by mental retardation and congenital heart defects. Children are susceptible to severe breathing problems and the development of pneumonia. In this regard, more than half of children do not live to see 2 years of age and only 10% can celebrate their 5th birthday. The cause of death is pneumonia, cardiac arrest or respiratory failure.
SMA 2 or Dubowitz disease
The disease is detected in children from 6 months to 1.5–2 years. Therefore, this form of SMA is often called late infantile. Typical for her:
- weakness and tremors in the muscles;
- tremor of fingers, tongue;
- stiffness of movement due to limited mobility of the limbs;
- developmental delay;
- underweight
Children with this diagnosis are able to sit, play, and eat independently, but cannot stand or move around. Unfortunately, the pathology tends to progress, which leads to a gradual weakening of the muscles of the chest and neck, resulting in the inability to hold the head straight and often it hangs limply. Then tendon reflexes disappear, the voice weakens and swallowing disturbances are noted.
Life expectancy with this diagnosis is about 10–12 years. But a third of patients die before the age of 4 years.
SMA 3 or Kugelberg-Welander disease
Spinal muscular atrophy of this type is usually diagnosed after 2 years. It is also manifested by muscle weakness, but not to the same extent as in SMA 1 or even SMA 2. Patients can stand independently, but only for a short period of time. Due to muscle atrophy, this is difficult for them.
Despite the existing disease, the child develops normally until the age of 10–12, which can mislead his family and raise doubts about the correctness of the diagnosis. But, reaching this time point, the first signs of SMA appear. The child begins to stumble more often than usual, falls and is unable to perform physical work or play sports, and often experiences fractures. Gradually, running and then walking become more and more difficult due to limited joint mobility. Subsequently, the teenager loses the ability to move without a wheelchair.
The progression of the pathology leads to the occurrence of severe scoliosis, which entails a change in the shape of the chest and the appearance of breathing difficulties. This is where the main threat of the disease to life lies.
SMA 4
This type of disease includes several different amyotrophies that do not affect life expectancy, but lead to disability:
- bulbospinal Kennedy;
- distal Duchenne-Aran;
- peroneal Vulpiana.
What they have in common is that the first clinical signs of the disease appear between 16 and 60 years of age, most often at 35–40 years of age. This is accompanied by extinction of tendon reflexes and noticeable muscle spasms. With Duchenne-Harand atrophy, the hands are most affected, and Vulpian disease is characterized by a change in the shape of the shoulder blades to pterygoid.
Nemaline myopathy
The second name for this disease is congenital non-progressive filamentous myopathy. Heredity is mainly transmitted in an autosomal dominant manner, but recessive and sporadic ones are also found. Possible death due to respiratory failure in early infancy. Severe skeletal pathologies are observed. The development of the disease may occur to varying degrees, or may not progress at all. In some cases, patients are forced to move using a sitting gurney, in others they suffer from respiratory failure. When diagnosing, a histological examination is carried out, which reveals unusual or rod-like, non-crimson bodies in the muscles. EMG usually confirms the diagnosis of myopathy.
Signs of dystrophy in children
Signs of dystrophy appear in young children depending on the form of the disease and its severity. Common symptoms are:
- excitation;
- worsening sleep;
- loss of appetite;
- fast fatiguability;
- weakness;
- growth retardation;
- weight loss.
For malnutrition of the I and II degrees (this term refers to an eating disorder accompanied by a lack of body weight) the following specific signs are characteristic:
- reduction in body weight up to 30%;
- loss of muscle tone;
- decreased tissue elasticity;
- thinning of subcutaneous tissue;
- vitamin deficiency;
- weakened immunity;
- pallor;
- bowel dysfunction (constipation and diarrhea alternate).
Hypotrophy of the third degree is characterized by more serious disorders in the child’s body:
- exhaustion;
- eyeballs sink;
- skin loses elasticity;
- breathing and heart rhythm are disrupted;
- blood pressure decreases;
- body temperature drops.
Paratrophy is expressed in such signs as:
- excess fat deposits;
- pallor;
- allergic;
- impaired intestinal function, dysbacteriosis;
- anemia;
- severe diaper rash.
Congenital structural myopathies
The second group is congenital structural myopathies (violations of the integrity of the cytoskeleton of muscle fibers and the occurrence of pathology in muscle biopsy). This group of diseases is characterized by a violation of the synthesis of proteins responsible for growth and other functions of muscle formation in the early development of the embryo.
Congenital structural myopathies include:
- central core disease;
- nemaline myopathy;
- centronuclear myopathy;
- megaconial myopathy;
- myopathy with disproportion of muscle fiber types;
- myopathy with multiple central cores;
- myotubular myopathy;
- myopathy with crystalline inclusions.
The clinical pictures of each of the diseases in this group are similar to each other and are characterized by muscle hypotonia and hypertrophy, decreased reflectivity in the tendons and increased concentrations of creatine phosphokinase in the blood. Slow progression is observed.