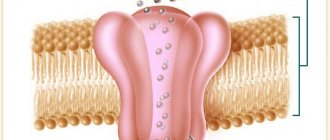
Myotonia is a rare genetic disease characterized by dysfunction of the muscular system. It occurs due to the malfunction of ion channels that transmit nerve impulses to muscle fibers. With myotonia, muscle contraction usually remains normal, but the relaxation phase is significantly prolonged, which leads to the appearance of characteristic symptoms.
The causes of the disease are purely genetic. The structure and functions of ion channels are encoded by specific genes: CNBP, DMPK and CLCN1. The occurrence of mutations in them leads to the development of myotonia.
Clinical manifestations
One of the classic symptoms of myotonia is the “fist” sign. If you ask the patient to quickly clench and unclench his fist, he will not be able to perform the last action. It will take some time to open your fingers. Likewise, the patient cannot quickly raise his eyelids, get out of bed or chair, or open his mouth. Other symptoms may include:
- stiffness,
- muscle hypertrophy,
- decreased muscle strength,
- diseases of the cardiovascular system,
- intellectual development disorder,
- impotence,
- hypersomnia.
There are several types of myotonia, which differ in a certain set of symptoms and the degree of their severity. In order to make an accurate diagnosis, the doctor is guided by the clinical manifestations of the disease, as well as information obtained during the diagnosis.
Reasons for the development of Rossolimo-Steinert-Kurshman myotonia
Rossolimo-Steinert-Kurshman myotonia is always a genetically transmitted defect. The key reason for its development is considered to be a violation in the DMPK gene, located on the nineteenth chromosome. The severity of the disease is directly proportional to the number of trinucleotide CTGs. Normally, this indicator is approximately 5-37, at 50-80 a mild form of the disease is observed, at 100-500 a late severe form is observed.
Under the influence of a violation of the DMPK gene in the body, a change occurs in myotonin protein kinase - a protein localized in skeletal and muscle tissue, in the myocardium, central nervous system, etc., which provokes the appearance of myotonic spasms, combined with atrophy of the muscles of the face, neck and limbs in the distal zones. In essence, hypertrophy of some muscle fibers occurs with simultaneous atrophy of others. As a result, some of these fibers are replaced either by other fibers or by fatty or connective tissues.
4.Treatment
Like most other rare diseases, Leiden-Thomsen disease is not well understood. Genetic engineering and genetic therapy methods are under development and are not yet used in clinical practice. Accordingly, therapy for diseases of this group is always palliative and is selected partly empirically, partly based on ideas about pathogenesis. To soften and reduce myotonic spasms, diacarb, diphenine, and calcium compounds (chloride or gluconate) are prescribed according to a specific regimen. Physiotherapy and physical therapy bring quite good results.
Opinions about the “natural” dynamics of congenital Leiden-Thomsen myotonia differ in different sources: one can find reports of both a steady slow progression throughout life and a tendency towards a spontaneous decrease in the severity of spastic-myotonic syndrome.
In any case, Leiden-Thomsen disease does not pose any threat to life, although, of course, it reduces the quality of life to one degree or another, causing psychological discomfort and obvious difficulties in social communication (which is a particularly painful problem at the manifest stage, i.e. in adolescence). Therefore, it is important to seek help in a timely manner, which will allow the disease to be diagnosed and adequate treatment prescribed.
Symptoms and signs of classical myotonia Rossolimo-Steinert-Curschmann
The very first signs of myotonia can appear as early as the age of 6-7 years, but most often become clearly visible in adolescence and young adulthood from 10 to 20 years. The list of these signs includes myopathy, cataracts, damage to the central nervous system and cardiovascular system, and endocrine disorders.
The main symptom is the presence of muscle spasms, which mainly affect the masseter facial muscle and the flexor muscle of the hand. Also, experts always note atrophy of various muscles, including the distal parts of all extremities, temporal, sternocleidomastoid, and facial muscles. Myopathic laryngeal paresis is observed with difficulty breathing, voice changes, deterioration of ventilation, sleep pathologies and aspiration and congestive pneumonia. Due to changes in muscle tissue and premature loss of tendon reflexes, most patients experience a change in gait.
In approximately 50% of cases, patients complain of a symptom such as arrhythmia, along with which bundle branch block, as well as hypertrophy of the left cardiac ventricle, can be observed.
On the part of the SNS, the disease causes hypersomnia, a decrease in intellectual abilities, and there are often cases when patients have a mild form of debility. Due to pathologies of the endocrine system, sexual dysfunction occurs. In young women, the changes consist of the manifestation of hirsutism, menstrual irregularities and early menopause, in young people - a decrease or absence of desire and impotence, cryptorchidism and hypogonadism.
Representatives of both sexes have a common symptom of Rossolimo-Steinert-Kurshman myotonia - changes in the structure of the hair with subsequent loss. In men, baldness occurs in the area of the temples and forehead; in women, a local focal or diffuse disorder occurs.
Symptoms of congenital Rossolimo-Steinert-Kurshman myotonia
The congenital type of myotonia can be noticed by a specialist even during the period of stay in the mother’s womb. This fetus does not show increased motor activity. Therefore, if a pregnant woman notices such behavior of the baby in the stomach, she should definitely undergo an ultrasound scan in the last three months before giving birth.
Signs of myotonia are also present in newborns. This is hypotonia of facial, masticatory muscles, distal areas of the limbs, and eyeballs. In addition, breathing disorders are also common. A little later, symptoms such as mental retardation and delayed motor development appear. The progression of these pathologies can occur at different rates. With the rapid pace of their development, the patient’s death from the disease can befall him at a very early age.
Clinical manifestations
A characteristic symptom of Thomsen's myotonia is the occurrence of a spasm in the skeletal muscles at the beginning of any action. It is worth noting that spasm never occurs at rest.
The first symptoms of the disease appear in children in infancy, but can be diagnosed much later (10-30 years). Young children begin to choke when crying and their voice changes. After crying, the facial muscles relax very slowly. However, an early symptom of the disease is the appearance of spasms in the hands. After clenching the hand into a fist and quickly trying to unclench it, the fingers and palm slowly relax and return to their normal position after a few seconds. With several repetitions of the “clenching and unclenching the hand into a fist” exercise, each subsequent action becomes easier and faster (myotonic phenomenon). The spasm is always maximally expressed at the beginning of a new action and gradually decreases after several repetitions, as the muscle “works into” the motor act.
There is no pain reaction in children in response to muscle contraction and, in general, the disease progresses favorably. Another symptom of the disease is muscle hypertrophy (enlargement). Muscle enlargement more often manifests itself in adulthood, but in some patients, already in childhood, upon examination, muscle hypertrophy is discovered, which is mistaken for the child’s athleticism. The athletic physique does not correspond to the actual picture of the muscle strength of the sick person - it is much lower. Hypothermia can trigger muscle contractions. As the disease progresses, the muscles of the tongue, masticatory group of muscles, muscles of the legs and trunk are involved. When quickly closing the eyes and trying to open them sharply, a person with myotonia cannot do this immediately; the eyelids open after a few seconds.
Children lag behind their peers in outdoor games and have difficulty running and jumping. Intellectual development does not suffer.
A neurological examination reveals percussion myotonia - when a muscle is hit with a hammer, it contracts in this area. For example, when hitting the tenor with a hammer, the thumb is quickly brought to the hand for several seconds. Percussion of the tongue provokes the formation of a “pit” in it - this is a distinctive symptom of this disease.
Diagnosis of Rossolimo-Steinert-Kurshman myotonia
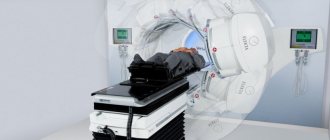
A specialist’s suspicion of this disease can be caused by a combination of any myotonic manifestations and dystrophic changes in muscle tissue that occur simultaneously with a lag in intellectual development, disturbances in the functioning of the endocrine and cardiovascular systems. The presence of all these signs in a patient requires confirmation of the diagnosis in the form of the results of a genealogical analysis, the results of which indicate an autosomal dominant type of genetic inheritance. In addition to these, DNA analysis data should be obtained for an accurate diagnosis. Additional studies are also needed: electromyography, electroneurography, electrocardiogram, hormonal analysis.
Determining an accurate diagnosis for suspected Rossolimo-Steinert-Kurshman myotonia requires a comprehensive study of the patient’s condition and the functioning of almost all of his organs. Therefore, specialists in genetics, cardiology, gynecology, andrology and endocrinologists can be involved in the diagnostic process.
When diagnosing, you need to pay attention not only to those changes that appear externally and are characteristic of Rossolimo-Steinert-Kurshman myotonia, but also to data on muscle biopsy and electromyographic studies.
During a biochemical analysis, the muscle enzyme and its level in tissues are studied, which is always elevated in this disease. A prerequisite for making an accurate diagnosis is the antenatal diagnosis of myotonia using amniocentesis.
Diagnostic options
If you have the above complaints, you should urgently visit a qualified specialist in the field of neurology. To confirm the conclusion, the doctor will prescribe certain clinical procedures, for example:
- Genealogical examination, which will show the presence of an autosomal dominant form of inheritance of the pathology.
- DNA test.
- Electromyographic diagnostics.
- Electroneurographic examination.
- Checking sex hormones.
- Electrocardiographic assessment.
In addition, consultations are held with doctors specializing in problems of hereditary, cardiological, endocrinological, gynecological and other types.
Differential diagnosis of Rossolimo-Steinert-Kurshman myotonia
It is necessary to differentiate Rossolimo-Steinert-Kurshman myotonia from other known types of myotonia, which in some situations may have similar signs and course. Atrophy of muscle tissue is characteristic exclusively of this disease and allows you to quickly distinguish it from Thomsen's myotonia, a sign of which is muscle hypertrophy. Early involvement of the facial muscles and dominant inheritance distinguish it from Becker myotonia. In addition to these conditions, it is necessary to differentiate Rossolimo-Steinert-Kurshman disease from ALS and Charcot-Marie-Tooth amyotrophy.
Prognosis and prevention
The prognosis for life with Thomsen's myotonia is favorable. However, it is impossible to get rid of myotonic attacks forever. But by taking a number of preventive measures, the patient is able to alleviate his condition and live a full life.
An important point is to determine the factors that provoke myotonic attacks. Most importantly, you should not overcool, you must avoid stressful situations, intense physical activity, sudden movements, prolonged stay in one position, and emotional shocks.
Treatment of Rossolimo-Steinert-Kurshman myotonia
In modern medical practice, there is currently no therapy designed to completely cure people of Rossolimo-Steinert-Curschmann myotonia. Patients diagnosed with this disease are prescribed a diet based on foods with minimal or no potassium levels. People suffering from this myotonia should not be overcooled, since low temperatures can provoke spasms at any time.
To reduce the risk of their sudden appearance and improve the general condition of the patient, he is prescribed the following drugs: quinine, novocainamide, diphenine simultaneously with diacarb. To these medications, experts recommend adding the mandatory use of anabolic steroids: nerobol, retabolil, methylandrostenediol. It is also necessary to include small doses of ATP and B vitamins in the list of required substances.
The above drugs have a good effect in both the classical and congenital types of the disease. All of these measures can reduce the negative effects of the disease on the patient’s body and prolong his life, but they are in no way able to completely rid him of myotonia and make him an absolutely healthy person again.