Причины
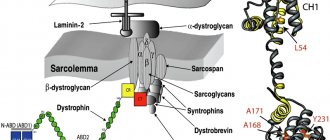
Заболевание наследственное, сцепленное с X-хромосомой, поэтому болеют практически всегда мальчики. Девочки являются носителем патологического гена (мальчики редко доживают до половозрелого возраста, к тому же, как правило, стерильны). В хромосоме происходит изменение структуры гена, отвечающего за синтез белка дистрофина.
Хоть содержание дистрофина в скелетной мускулатуре предельно мало (тысячные доли процента), без него быстро развивается некроз мышечной ткани, развивается прогрессирующая дистрофия мышц. Если ген повреждается на участке, полностью разрушающем синтез белка-дистрофина, развивается дистрофия Дюшенна. При вовлечении в процесс малозначимых отделов белка, заболевание принимает форму дистрофии Беккера.
Мышечные дистрофии двусторонние
В начальный период может преобладать миодистрофия с одной стороны, но по мере развития заболевания степень поражения становится одинаковой в симметричных мышцах больного. Со временем течения болезни практически во всех мышцах снижается их мышечная сила. На теле больного, страдающего мышечной дистрофией, появляются участки гипертрофических мышц. Это псевдогипертрофия, которая не связана с увеличением мышечных волокон. Псевдогипертрофия мышц связана с отеком в мышцах ног или рук. Такие мышцы плотные, но слабые.
Симптомы
Начало развития симптомов начинается в раннем детском возрасте, чаще от 1 до 3 лет. Изначально отмечается отставание в моторном развитии, ребенок поздно начинает ходить, часто спотыкается при ходьбе, быстро устает. Позже развивается постоянная патологическая утомляемость мышц. Ребенок практически не может взбираться по лестнице. Походка начинает напоминать «утиную».
Характерным симптомом является симптом «лесенки»: попытка встать из положения сидя происходит с использованием рук, постепенно, медленно, в несколько этапов.
Постепенно начинает отмечаться атрофия мышц, вначале проксимальных отделов нижних, потом верхних конечностей. Позже атрофируются мышцы тазового пояса, бедер, спины, плечевого пояса. Почти всегда развивается «осиная» талия, искривление позвоночника, выпирание лопаток (крыловидные лопатки).
Практически всегда имеет место характерный симптом прогрессирующей мышечной дистрофии Дюшенна – псевдогипертрофия мышц голеней. Мышцы, хоть и увеличены в объеме, они не имеют достаточной силы, очень болезненны на ощупь.
Можно выделить три стадии заболевания: — I стадия – слабость проявляется лишь при значимой физической нагрузке (обычно первый год течения болезни). — II стадия – затруднен подъем по лестнице, быстро развивается слабость при ходьбе. — III стадия – представляет собой параличи, контрактуры мышц с невозможностью cсамостоятельного передвижения.
По видам течения подразделяется на: Быстрое прогрессирование. Способность к передвижению утрачивается быстро, в течение первых 4-5 лет с начала заболевания. Средний темп прогрессирования: передвигаться пациент не может спустя 10 лет. Медленное прогрессирование: выраженных двигательных нарушений через 10 лет от начала болезни нет. Обычно такой вариант характерен для других типов мышечных дистрофий, нежели дистрофии Дюшенна.
Лечение спинально-мышечная атрофия
Больным назначается комплексная консервативная терапия, направленная на улучшение способности нервных импульсов проходить к мышцам и работы головного мозга. В этих целях рекомендуется прием:
- ноотропов;
- препаратов α-липоевой кислоты, ацетил-L-карнитина, α-глицерофосфохолина;
- витаминных комплексов, включающих, прежде всего, витамины группы В;
- средств, улучшающих обмен веществ.
Сегодня в разработке находятся специфические лекарственные средства, способные воздействовать на причину развития СМА – дефицит ряда белков. Но в данный момент они находятся на стадии испытаний. Пока что единственным способом хотя бы частично обеспечить организм необходимыми белками является соблюдение специальной диеты. Она подразумевает употребление продуктов, богатых на аминокислоты, а именно зерновых культур, орехов, кисломолочных продуктов, рыбы, мяса. Весьма полезно включение в меню шпината, брокколи, грейпфрутов. Особенно ценны блюда из бурого риса и овса.
Для поддержания мышечного тонуса рекомендованы:
- занятия ЛФК;
- массаж;
- физиотерапевтическое лечение;
- нейромышечная стимуляция.
Диагностика
Клиническая картина очень яркая. Часто заболевание ставится после выяснения генетического анамнеза (наличие случаев в семье), неврологического осмотра. В неврологическом статусе отмечается пропадание коленных рефлексов, чуть позже исчезают рефлексы с бицепса, трицепса. Ахилловы рефлексы долгое время сохранны.
Внешне может выявиться деформация суставов стопы, имеются признаки кардиомиопатии: нарушение пульса, глухость сердечных тонов, расширение полостей сердца по ЭхоКГ, изменения на электрокардиограмме.
Важным фактором является повышение биохимических показателей креатинфосфокиназы (фермент-показатель распада мышц). Активность данного фермента увеличивается в десятки раз. Имеется прямая корреляция между степенью увеличения активности фермента и выраженностью проявлений дистрофии Дюшенна. [!] В сложных диагностических ситуациях проводят цитологическое исследование.
Патологоанатомическая картина заболевания
Посмотрим, что происходит внутри мышечных клеток у пациентов, страдающих миодистрофией Дюшенна. Для этого сделаем надрез кожи, расширим расширителем и возьмем небольшой кусочек мышечных волокон.
На фото: биопсия мышечной ткани
На фото: биопсия мышечной ткани
Типичный признак миодистрофии в первую очередь – это разный диаметр мышечных волокон. У здорового человека диаметр мышечных волокон одинаковый.
Характерные признаки мышечной дистрофии – атрофированные и гипертрофированные волокна, множественные внутренние ядра и отек.
Изучая окрашенные срезы скелетной мышцы, я увидел денервацию миофибрилл, значительный разброс в размере миофибрилов и выраженный отек.
Пояснение к первому фото:
- Бледно-фиолетового цвета – это мышечные волокна в разрезе.
- Светлые пятна как внутри волокон, так и снаружи – это отек.
- Темные точки – это ядра, которые отек сместил к периферии.
На втором фото показано нормальное мышечное волокно здорового человека.
Степень тяжести мышечной дистрофии по данным электронной микроскопии ориентируется на следующие показатели:
- при легкой степени разница в размере мышечных волокон выражена умерено, начальные признаки отека (белый цвет).
На фото: биопсия мышечных волокон при легкой (A), средней (B) и тяжелой степени дистрофии (C).
- средняя степень тяжести соответствует перемещению ядер в центр мышечных волокон, расширению межфибриллярного пространства из-за увеличения отека между клетками.
На фото: мышечные волокна при прогрессирующей мышечной дистрофии средней степени тяжести:
а) светло-фиолетового цвета мышечные волокна;
б) светлые пятна внутри мышечных волокон – отек, оттолкнувший ядра из центра клетки к периферии;
в) темные точки – ядра мышечных клеток;
г) стрелкой показана мышечная клетка, которая не может двигаться из-за уменьшения обменных процессов, – темнеет в сторону фиолетового цвета.
- тяжелая степень характеризуется обширными очагами деструкции миофибрилл, их фрагментация и дезорганизация, появление гиалиноподобного вещества и отека между мышечными клетками. Функционально такая ткань обладает слабой силой, быстро наступает утомление и развиваются признаки мышечной усталости. Фото будет представлено немного ниже.
Вот такое состояние мышц было у Эмине до обращения ко мне:
Пояснение к фото “тяжелая степень мышечной дистрофии”:
- Мышечные волокна в разрезе окрашены в синий цвет.
- Красные точки – это ядра мышечных клеток.
- Отек – неокрашенный белый цвет.
Лечение и прогноз жизни
Лечение симптоматическое. Используются гормональные препараты для остановки разрушения мышечного волокна, фосфолипиды в качестве защиты клеток мышц от разрушения, элементы лечебной гимнастики. Внедряются в практику различные ортопедические приспособления для облегчения передвижения. Массаж строго противопоказан в большинстве случаев, так как может приводить к ускорению распада мышц. Лечение наследственных заболеваний – дело будущего.
Прогноз жизни для пациентов неблагоприятный. Течение заболевание прогрессирующее. Неизбежен летальный исход. Как правило, к семилетнему возрасту развивается выраженная симптоматика, приводящая к 13-14 годам к полной обездвиженности. Больные редко доживают до 18-20 лет.
Что такое спинальная мышечная атрофия и ее виды
Под этим термином объединяется несколько различных видов наследственных заболеваний, сопровождающихся ограничением двигательных способностей. Этим и объясняется тот факт, что в части случаев нарушения обнаруживаются не в младенческом возрасте, а у подростков или уже зрелых людей.
Впервые заболевание было описано в 1891 г. Г. Верднигом и в 1892 г. было выделено в отдельную нозологическую единицу Дж. Хоффманом, благодаря стараниям которых и получила свое второе название. Примерно через полвека Е. Кугелбергом и Л. Веландером была открыта другая подобная болезнь, развивающаяся в более позднем возрасте и отличающаяся более благоприятным течением.
Различают следующие формы патологии:
- СМА 0;
- СМА 1 (тяжелая форма);
- СМА 2 (промежуточная форма);
- СМА 3 (легкая форма);
- СМА 4 (поздняя форма).
Причина спинально-мышечная атрофии у детей
Все их объединяет то, что причина их возникновения кроется в мутации рецессивного гена 5 хромосомы SMN. Это приводит к сбоям в продукции протеинов в организме, являющихся строительным материалом всех клеток. В результате страдают мотонейроны спинного мозга и постепенно разрушаются. Поскольку без них невозможна передача нервных импульсов к мышечным волокнам, они постепенно атрофируются, что становится причиной утраты способности двигаться.
К счастью, даже при наличии у обоих родителей мутации гена SMNу них с 75% вероятностью может родиться здоровый ребенок. Но практически всегда он также будет носителем этого гена. Поэтому при планировании беременности стоит проходить генетическое исследование, особенно при наличии случаев СМА в семье.
СМА 0
Это врожденная болезнь, признаки которой обнаруживаются обычно еще в роддоме. Она встречается редко и ее часто объединяются со СМА-1. Для этого вида типично абсолютное отсутствие подвижности, слабость мышц, отсутствие сухожильных рефлексов и ограничение функциональности коленных суставов. С первых дней жизни ребенок страдает от нарушения дыхания.
Спинально-мышечную атрофию важно дифференцировать с перинатальной энцефалопатией и родовыми травмами, но если при них состояние детей постепенно улучшается, то при СМА оно не меняется. Более того часто присоединяются осложнения, которые практически всегда приводят к смерти младенцев в течение первого месяца жизни.
СМА 1 или болезнь Вердинга-Гоффмана
Этот тип течения спинальной мышечной атрофии характеризуется очень тяжелым протеканием. Обычно она обнаруживается до 6-ти месяцев и сопровождается слабостью мышц, периодическими спазмами, что сложно заметить в связи с особенностями анатомии детей первого года жизни (присутствия ярко-выраженной подкожно-жировой клетчатки).
Также заболевание проявляется регулярно пробегающей по языку дрожью, снижением рвотного, сосательного, глотательного рефлексов. Это приводит к возникновению серьезных трудностей при кормлении. Присутствует нарушение слюноотделения, кашель. Ребенок часто громко кричит.
Поскольку мышцы груди развиты недостаточно, можно заметить, что форма грудной клетки более плоская. Кроме того, дети с такой патологией лежат и спят в позе «лягушонка»: с отведенными в стороны плечами и бедрами при согнутых в коленях и локтях конечностях. Они способны к полугоду научиться держать голову (она часто имеет меньшие размеры, чем у здоровых детей), но им не под силу самостоятельно сидеть или принимать вертикальное положение тела.
Эта форма спинально-мышечной атрофии может сопровождаться олигофренией и врожденными пороками сердца. Дети подвержены тяжелым нарушениям дыхания, развитию воспаления легких. В связи с этим более половины детей не доживает до 2 лет и только 10% могут отметить свой 5-летний юбилей. Причиной смерти становятся пневмония, остановка сердца или дыхательная недостаточность.
СМА 2 или болезнь Дубовица
Заболевание обнаруживается у детей от 6 месяцев до 1,5–2 лет. Поэтому такую форму СМА часто называют поздней младенческой. Для нее типично:
- слабость и дрожь в мышцах;
- тремор пальцев, языка;
- скованность движений, обусловленная ограничением подвижности конечностей;
- задержка развития;
- недобор веса.
Дети с таким диагнозом способны самостоятельно сидеть, играть, есть, но стоять и передвигаться нет. К сожалению, патология склонна прогрессировать, что приводит к постепенному ослаблению мышц груди и шеи, следствием чего становится невозможность удерживать голову прямо и часто она безвольно свисает. Затем пропадают сухожильные рефлексы, слабеет голос и отмечаются нарушения акта глотания.
Длительность жизни при таком диагнозе составляет около 10–12 лет. Но треть больных погибает в возрасте до 4-х лет.
СМА 3 или болезнь Кугельберга-Веландера
Спинальную мышечную атрофию этого вида диагностируют обычно после 2 лет. Она так же проявляется слабостью мышц, но не в такой степени как при СМА 1 или даже СМА 2. Больные могут самостоятельно стоять, но только в течение короткого периода времени. В связи с атрофией мышц это дается им с трудом.
Несмотря на имеющееся заболевание, до 10–12 лет ребенок развивается нормально, что может ввести его родных в заблуждение и вызвать сомнения в правильности поставленного диагноза. Но, достигая этого временного рубежа, возникают первые признаки СМА. Ребенок начинает спотыкаться чаще обычного, падает и не может выполнять физическую работу или заниматься спортом, часто сталкивается с переломами. Постепенно бег, а затем и ходьба даются все сложнее из-за возникновения ограничения подвижности суставов. Впоследствии подросток теряет способность передвигаться без инвалидного кресла.
Прогрессирование патологии приводит к возникновению тяжелого сколиоза, что влечет за собой изменение формы грудной клетки и появление трудностей при дыхании. Именно в этом таится главная угроза болезни для жизни.
СМА 4
К этому типу заболевания относят несколько разных не влияющих на продолжительность жизни, но приводящих к инвалидизации амиотрофий:
- бульбоспинальную Кеннеди;
- дистальную Дюшена-Арана;
- перонеальную Вюльпиана.
Их объединяет то, что первые клинические признаки заболевания проявляются в период от 16 до 60 лет, чаще в 35–40 лет. Это сопровождается угасанием сухожильных рефлексов и заметными спазмами мышц. При атрофии Дюшена-Арана сильнее всего страдают кисти, а для болезни Вюльпиана характерно изменение формы лопаток на крыловидную.
Немалиновая миопатия
Второе название данного заболевания — врожденная непрогрессирующая нитеобразная миопатия. Наследственность в основном передается по аутосомно-доминантному типу, но встречается также рецессивный и спорадический. Возможен летальный исход вследствие дыхательной недостаточности в раннем младенческом возрасте. Наблюдается сильно выраженные патологии скелета. Развитие болезни может происходить в той или иной степени, а может не прогрессировать вовсе. В отдельных случаях больные вынуждены передвигаться с помощью сидячей каталки, в других — страдают от дыхательной недостаточности. При диагностировании проводится гистологическое исследование, которое выявляет в мышцах неподобные или палочкоподобныене малиновые тельца. ЭМГ обычно утверждает диагноз миопатии.
Признаки дистрофии у детей
Проявляются признаки дистрофии у детей раннего возраста в зависимости от формы заболевания и его степени тяжести. Общими симптомами считаются:
- возбуждение;
- ухудшение сна;
- потеря аппетита;
- быстрая утомляемость;
- слабость;
- задержка роста;
- потеря веса.
Для гипотрофии I и II степени (под таким термином имеется в виду расстройство питания, сопровождающееся дефицитом массы тела) характерны такие специфические признаки, как:
- снижение массы тела до 30 %;
- потеря мышечного тонуса;
- снижение эластичности тканей;
- истончение подкожной клетчатки;
- витаминная недостаточность;
- ослабление иммунитета;
- бледность;
- нарушение стула (запоры и поносы чередуются).
Гипотрофия III степени отличается более серьёзными нарушениями в детском организме:
- истощение;
- глазные яблоки западают;
- кожа теряет эластичность;
- дыхание и сердечный ритм нарушаются;
- артериальное давление снижается;
- температура тела падает.
Паратрофия выражается в таких признаках, как:
- избыточные жировые отложения;
- бледность;
- аллергичность;
- нарушенная работа кишечника, дисбактериоз;
- анемия;
- сильные опрелости.
Врожденные структурные миопатии
Вторая группа — врожденные структурные миопатии (нарушения целостности цитоскелета мышечных волокон и возникновение патологии в биоптате мышц). Эта группа заболеваний характеризуется нарушением синтеза белков, отвечающих за рост и другие функции формирования мышц в раннем развитии эмбриона.
К врожденным структурным миопатиям относят:
- болезнь центрального стержня;
- немалиновую миопатию;
- центронуклеарную миопатию;
- мегакониальную миопатию;
- миопатию с диспропорцией типов мышечных волокон;
- миопатию со множественными центральными стержнями;
- миотубулярную миопатию;
- миопатию с кристаллическими включениями.
Клинические картины каждого из заболеваний данной группы похожи друг на друга и характеризуются мышечной гипотонией и гипертрофией, пониженной рефлективностью в сухожильях и повышением концентрации в крови креатинфосфокиназы. Наблюдается медленное прогрессирование.