Особенности течения мышечной дистрофии Ландузи-Дежерина
Нервно-мышечное заболевание характеризируется прогрессирующим поражением лицевой мускулатуры, плеч и лопаток. Также поражаются мышцы других частей тела, но значительно медленнее и менее выражено. Особенностью течения является несимметричность деструктивного процесса. Зачастую первыми атрофируются мышцы плечевого пояса, постепенно распространяясь на мимическую мускулатуру, приводя к амимичности лица, слабости и невозможности поднять руки.
В отличие от других миодистрофий, при миопатии Ландузи-Дежерина занятия спортом и физические нагрузки не замедляют патологический процесс, а наоборот ускоряют деструкцию мышечной ткани. В дальнейшем отмечается нарушение сердечной деятельности, может произойти потеря зрения и снизиться слух.
Впервые данную миопатию описали французские врачи Ландузи и Дежерин в 1884 году.
Известно, что диагноз является генетически обусловленным, с аутосомно-доминантным типом наследования. У большинства больных обнаруживаются генные мутации, но у некоторых пациентов генетические нарушения не выявляются.
Плече-лопаточно-лицевая миопатия Ландузи-Дежерина относится к редким патологиям, с частотой заболеваемости 1-1,5 на 100 000 населения. Из-за наследственной предрасположенности она часто выявляется у кровных родственников. Первые симптомы возникают еще в детском возрасте. Пик заболеваемости и выраженности клинических проявления – переходной возраст, период беременности и климактерический период.
Патологию продолжают изучать, но причина возникновения дефекта и развития атрофии не известна. Патогенетического лечения в настоящее время не существует, из-за этого болезнь продолжает относиться к тяжелым заболеваниям. Несмотря на то, что длительное время пациенты остаются трудоспособными и могут самостоятельно себя обслуживать, в большинстве случаев диагноз приводит к их инвалидизации.
Психотерапия
Начало клинических проявлений и последующие ухудшения, как правило, провоцируются психическим стрессом. Более того, тяжелое заболевание мышц, само по себе, серьезный психический стресс. На фоне болезни часто развиваются вторичные депрессии с апатией и нежеланием заниматься своим здоровьем. А это ведет к прогрессированию болезни и возможной гибели пациента.
Если это нужно, мы проводим нашим пациентам курс психотерапевтического лечения. Наши пациенты становятся более устойчивыми к психическим раздражителям, становятся активнее, позитивно относятся к лечению, и это незамедлительно сказывается на работе мышц.
Кроме того, мы обучаем пациентов специальным психотехникам для работы с собственными мышцами.
При наличии признаков депрессии возможно назначение современных антидепрессантов (селективные ингибиторы обратного захвата серотонина, такие как Ципралекс, Флуоксетин и др.). Эти антидепрессанты не снижают, а повышают активность пациента и настроение, не вызывают мышечной слабости.
Причины развития
Первичные причины возникновения биохимических дефектов, приводящих к миопатии, неизвестны. Но, анализируя статистические данные, ученые выделяют несколько факторов развития миопатии:
- наследственный фактор – часто страдают члены одной семьи;
- генетическая предрасположенность – у многих выявляется дефект 4 хромосомы;
- вирусные заболевания у матери во время беременности;
- патология беременности у матери.
У некоторых пациентов миопатия возникает на фоне нормального здоровья без видимых на то причин. В таком случае диагноз классифицируют как идиопатическую миопатию.
Из-за отсутствия информации о начальных процессах развития патологии врачам практически не удается их остановить и восстановить утраченные функции организма.
Симптомы
Первые признаки заболевания возникают в 10-20 лет, когда отмечается чрезмерная мышечная слабость, быстрая утомляемость и медленное развитие мускулатуры плечевого пояса. Больным трудно поднимать руки, выполнять физические упражнения. Из-за атрофии мышечной ткани у них чрезмерно вступают лопатки, приобретая крыловидную форму, промежуток между ними расширен, грудная клетка уплощенная. Слабость мускулатуры способствует развитию сколиоза.
Дистрофия мышц плеча, грудной клетки и трапециевидной мышц приводит к развитию симптома свисающего плеча. Частым осложнением в таком случае являются вывихи плечевого сустава. У больных возникают трудности в повседневной жизни и в быту. Им сложно расчесывать волосы, умываться, чистить зубы. Из-за привычных ранее нагрузок они чрезмерно быстро устают, вплоть до невозможности их выполнения.
Прогрессируя, миопатия вовлекает в процесс лицевые мышцы. Лицо становится амимичным. Из-за поражения круговых мышц рта и глаз сложно либо невозможно зажмурить глаза, сжать губы. Это приводит к частым воспалениям глаз, травмированию роговицы.
Распространенной жалобой является нарушение речи – неразборчивая, медленная. Это значительно ухудшает качество жизни, особенно социальной жизни и трудовой деятельности. Также трудности возникают во время приема пищи. Больным тяжело есть твердую пищу, пережевывать ее и глотать.
Характерными признаками болезни являются: вывороченные губы «губы тапира», поперечная улыбка «лицо Джоконды», полированного лба.
В дальнейшем возможна прогрессирующая мышечная дистрофия в других частях тела. При поражении тканей ягодиц и бедра, мышечная слабость способствует чрезмерной утомляемости от ходьбы, которая может привести к хромоте. Поражение икроножных мышц сопровождается симптомом свисающей стопы, выраженной слабостью при ходьбе и невозможностью бегать.
В зависимости от того, какие симптомы более выражены и какова очередность их возникновения, выделяют следующие формы миопатии Ландузи-Дежерина:
- лицелопаточно-плечевая;
- лицелопаточная-плече-перонеальная;
- лицелопаточно-плече-ягоднично-бедренная;
- лицелопаточно-плече-ягоднично-бедренная-перонеальная;
- лицелопаточно-плече-перонеально- ягоднично-бедренная;
- инфантильная форма.
Наиболее тяжелой является инфантильная форма. Она возникает у детей до 5 лет. Характеризируется симметрическим парезом лицевых мышц, поражением плеч, грудной клетки, отсутствием сухожильных рефлексов. Из-за стремительно нарастающей слабости у детей возникает дыхательная недостаточность вплоть до необходимости в искусственной вентиляции легких.
Важно! Часто родители детей с чрезмерной мышечной слабостью стараются восстановить организм с помощью спортивных секций и физической нагрузки. В результате этого болезнь прогрессирует еще быстрее.
Журнал «Здоровье ребенка» 1 (28) 2011
Лицелопаточно-плечевая миодистрофия Ландузи — Дежерина была описана Ландузи и Дежериным в 1884 г. Большинство отечественных детских неврологов впервые познакомились с данной патологией в руководстве Л.О. Бадаляна [1], которое до настоящего времени многим специалистам служит настольной книгой. При этом следует отметить и значительный вклад коллектива ученых, которые многие годы занимались и занимаются изучением диагностики, механизмов развития, клинических проявлений и лечения нейромышечных заболеваний [2, 5].
Миодистрофия Ландузи — Дежерина наследуется по аутосомно-доминантному типу с высокой пенетрантностью и встречается с частотой 0,9–2 на 100 000 населения. Установлена генетическая гетерогенность лицелопаточно-плечевой миодистрофии: в 90–95 % семей обнаруживается сцепление с локусом 4q35 (1A тип болезни), а в остальных 5–10 % — с локусом 10q26 (IB тип). Первичные биохимические дефекты в настоящее время неизвестны [4, 8, 11].
В классическом варианте первые признаки заболевания появляются в основном в возрасте 10–20 лет. Атрофии и мышечная слабость локализуются в области мимической мускулатуры, лопаток и плеч. Сначала атрофии наблюдаются в плечевом поясе, с последующим распространением на лицо. Обычно начальными проявлениями становятся затруднение подъема рук над головой, выступающие «крыловидные» лопатки и сколиоз. При прогрессировании процесса грубо страдают круговые мышцы рта и глаз — не удается крепко зажмурить глаза и сжать губы. Вследствие атрофии лицо становится гипомимичным. Как правило, больные сами отмечают изменение своей мимики, их речь становится неразборчивой. Следует отметить и характерные симптомы в виде поперечной улыбки («улыбка Джоконды»), вывороченных губ («губы тапира»), «полированного» лба. Атрофии двуглавой и трехглавой мышц плеча, большой грудной, передней зубчатой, трапециевидной мышц могут обусловливать возникновение симптомов свободных надплечий, «крыловидных» лопаток, появление широкого межлопаточного промежутка, уплощения грудной клетки и сколиоза. В ряде случаев атрофии распространяются на мышцы ног. При этом слабость наиболее заметна в группе малоберцовых мышц по свисающей стопе, но может быть и в проксимальных отделах ног [5, 6, 10]. В зависимости от последовательности и характера распространения процесса выделяют собственно лицелопаточно-плечевую форму, лицелопаточно-плече-перонеальную, лицелопаточно-плече-ягодично-бедренную, лицелопаточно-плече-ягодично-бедренно-перонеальную и лицелопаточно-плече-перонеально-ягодично-бедренную формы. На ранних стадиях болезни мышечный тонус снижен в проксимальных группах мышц, глубокие рефлексы снижены преимущественно с двуглавой и трехглавой мышц плеча [1].
Необходимо отметить асимметричность атрофии, что является характерной клинической особенностьюданной патологии. В некоторых случаях имеет место псевдогипертрофия мышц. Контрактуры и ретракции выражены умеренно. Кардиомиопатия бывает в редких случаях. При проведении ангиоретинографии могут выявляться аномалии сосудов сетчатки. Во многих случаях при тяжелых глазных проявлениях находят телеангиэктазии, отек и отслойку сетчатки. Коагуляция телеангиэктазий предотвращает развитие слепоты. Может наблюдаться также и снижение слуха. Вышеперечисленные симптомы рассматриваются в качестве составляющей части фенотипических проявлений данной патологии. Тип течения болезни в большинстве случаев относительно благоприятный. Особо мы хотим акцентировать внимание на том, что физические перегрузки, интенсивные спортивные занятия и нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни [9].
Несмотря на определенные успехи молекулярной генетики и другие достижения медицинской науки, диагностика данного заболевания на современном этапе основывается прежде всего на особенностях клиники (преимущественно лицелопаточно-плечевая локализация миодистрофического процесса) и на генеалогическом анализе (аутосомно-доминантный тип наследования).
Дифференциальную диагностику следует проводить с другими прогрессирующими мышечными дистрофиями (Эрба — Рота, Беккера и т.д.) и с вторичными миопатическими синдромами, которые возникают на фоне воспалительных, сосудистых, токсических и метаболических процессов.
Для уточнения диагноза используют биохимическое исследование с определением креатинфосфокиназы (КФК), электронейромиографию (ЭНМГ) и биопсию мышц. Уровень КФК может повышаться в 5 раз, но в некоторых случаях содержание фермента нормальное. На ЭНМГ регистрируются как миопатические, так и денервационные потенциалы. При проведении гистологических исследований во многих мышцах конечностей выявляют минимальные изменения, наибольшее число патологических признаков отмечается в надлопаточных мышцах, где обнаруживаются явления прогрессирующей дегенерации и небольшой краевой денервации [5, 7, 9].
Хотим обратить внимание на то, что в монографиях зарубежных авторов последних лет описана и инфантильная форма лицелопаточно-плечевой миодистрофии, которая быстро прогрессирует и приводит к тяжелой инвалидизации. Симптомы возникают еще в грудном возрасте, но не позднее 5 лет жизни в виде двустороннего пареза мимических мышц, что может имитировать врожденное поражение лицевых нервов. В дальнейшем развиваются ринолалия, а иногда птоз. Прогрессирующая проксимальная мышечная слабость возникает спустя 1–2 года после дебюта и в первую очередь захватывает плечи, а затем мышцы таза. Может отмечаться и псевдогипертрофия голеней. Сухожильные рефлексы снижены, а со временем исчезают. Отмечается быстрое нарастание слабости, что приводит к летальному исходу вследствие дыхательной недостаточности до достижения пациентом 20-летнего возраста. Очень редко в течение длительного срока слабость не прогрессирует, а тяжелая инвалидизация не наступает вплоть до зрелого возраста. У половины родственников больных обнаруживаются телеангиэктазии сетчатки и высокая частота снижения слуха. Диагноз данной патологии должен быть заподозрен у каждого ребенка с прогрессирующей лицевой диплегией, но в каждом случае необходимо исключить миастению и глиому мозгового ствола [8, 10].
Приводим таблицу, в которой представлены данные по локализации генов, ответственных за наследственные миопатии, с указанием первичных биохимических дефектов и краткой функциональной характеристикой этих белков (табл. 1) [12]. Как было отмечено выше, первичные биохимические дефекты в случае лицелопаточно-плечевой миодистрофии Ландузи — Дежерина в настоящее время неизвестны.
В настоящее время терапевтические возможности при миодистрофии Ландузи — Дежерина весьма ограниченны. Симптоматическое лечение направлено прежде всего на предотвращение развития контрактур, поддержание имеющейся мышечной силы и на снижение скорости развития атрофии. Основная задача состоит в том, чтобы максимально продлить период, в течение которого больной способен самостоятельно передвигаться, так как в лежачем положении быстро нарастают контрактуры, сколиоз, дыхательные расстройства. Терапевтический комплекс должен включать в себя лечебную гимнастику, массаж, ортопедические мероприятия, медикаментозную терапию, сбалансированное питание. В медикаментозном лечении прогрессирующих мышечных дистрофий следует отметить назначение курсами препаратов метаболического действия, направленных на улучшение и поддержание обменных, энергетических процессов неповрежденных миоцитов, кардиомиоцитов (кардонат, элькар, витамин Е, метионин, цитофлавин, АТФ-лонг, милдронат, кокарбоксилаза). Определенное значение имеет коррекция питания пациента. Рекомендуется диета с высоким содержанием белка и низким содержанием жиров, с пониженной калорийностью при оптимальном содержании витаминов и микроэлементов [5, 6, 8]. Важную роль играет и психологическая поддержка больного, продолжение обучения, правильная профессиональная ориентация.
В клинике детской психоневрологии ГУ «ИПАГ АМН Украины», где наряду с распространенными тяжелыми и труднодиагностируемыми неврологическими заболеваниями у детей также концентрируются и редкие неврологические нозологии, в течение последних пяти лет наблюдалось 7 пациентов с прогрессирующей миодистрофией Ландузи — Дежерина. Приводим описание истории болезни пациента, проходившего обследование и лечение в нашем отделении. Считаем, что этот клинический случай представляет значительный интерес, учитывая отягощенный семейный анамнез по данному заболеванию (11 случаев заболевания в роду).
Больной М., 16 лет, поступил в отделение с жалобами на слабость в верхних конечностях — невозможность поднять руки вверх. Слабость в руках больной отмечает на протяжении года, в то время как мать, страдающая аналогичным заболеванием, заметила снижение силы в верхних конечностях сына года три назад. Слабость постепенно нарастает. Заметное ухудшение отмечали после физических нагрузок — спортивных занятий на турнике и подъема тяжестей.
Из анамнеза жизни: мальчик от 2-й нормально протекавшей беременности, 2-х родов со слабостью родовой деятельности, родился с массой 4000 г, длиной 58 см. Развивался соответственно возрасту, учится в 9-м классе, удовлетворительно.
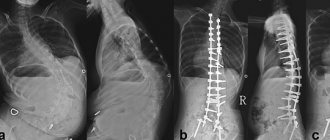
Неврологический статус: интеллект сохранен. Функция глазодвигательных нервов не нарушена. Выражена гипомимия лица и гипотрофия мышц плечевого пояса, «крыловидные» лопатки (рис. 1–3). Мышечная сила в проксимальных отделах верхних конечностей снижена до 3 баллов, больше слева, в дистальных — 5 баллов, в нижних конечностях — 3 балла. Мышечный тонус понижен. Сухожильные рефлексы на верхних и нижних конечностях живые, равномерные; брюшные рефлексы живые. Патологических стопных знаков нет. Походка не изменена. Координаторные пробы выполняет четко.
Обследование: общеклинические анализы без патологических изменений; активность ферментов сыворотки крови — АЛТ — 23 ед/л (N — 8–40 ед/л), АСТ — 34 ед/л (N — 10–41 ед/л), КФК — 148 (N < 270 ед/л); ЭНМГ — признаки первично-мышечного поражения.
Консультирован генетиком: подтвержден диагноз прогрессирующей мышечной дистрофии Ландузи — Дежерина.
Семейный анамнез: у матери пробанда, 41 года, в 14 лет началось заболевание, проявившееся слабостью мышц плечевого пояса; слабость мимической мускулатуры появилась с 35–36 лет. Аналогичное заболевание отмечается у ее отца, у 5 из 10 родных братьев и сестер и у 4 племянников (рис. 4). По месту жительства женщина получает социальное пособие в связи с прогрессирующей мышечной дистрофией Эрба — Рота.
Анализируя семейный анамнез, обращаем внимание на то, что заболевание имеет наследственный характер с аутосомно-доминантным типом наследования. Ювенильная конечностно-поясная миодистрофия Эрба — Рота наследуется по аутосомно-рецессивному типу. Отличием клиники миодистрофии Эрба — Рота является преимущественно восходящий (начинающийся с нижних конечностей), а не нисходящий характер поражения, как наблюдается у всех больных членов описанной семьи. Поэтому считаем, что у мамы мальчика правомочен диагноз не миодистрофии Эрба — Рота, а миодистрофии Ландузи — Дежерина.
Представленный нами случай демонстрирует редкую форму прогрессирующей мышечной дистрофии с аутосомно-доминантным типом наследования — лицелопаточно-плечевую миодистрофию Ландузи — Дежерина. Особенностью случая является отягощенный семейный анамнез по данному заболеванию (11 больных в роду), что, однако, не вызвало настороженности медицинских работников при появлении первых симптомов у пациента. Важно отметить, что в случае своевременной диагностики и необходимого ограничения физических нагрузок возможно было бы избежать быстрого прогрессирования заболевания.
Таким образом, мышечные дистрофии характеризуются прогрессирующей мышечной слабостью, атрофией мышц и двигательными нарушениями. Однако различные формы миопатий отличаются разным типом наследования, вариабельностью возраста начала заболевания, преимущественной локализацией поражения мышц и другими признаками, что необходимо уметь своевременно диагностировать и дифференцировать. Очевидно, что в дальнейшем успех в лечении прогрессирующих мышечных дистрофий будет зависеть от новейших разработок в области молекулярной генетики, генной терапии. Поэтому вовремя назначенное лечение позволит предотвратить быстрое прогрессирование заболевания, улучшить качество жизни пациента и увеличить продолжительность его жизни.
В заключение хотим отметить, что публикация в широко читаемом педиатрическом журнале статьи, посвященной сугубо неврологической патологии, обусловлена тем, что больные первично обращаются не к неврологу, а к детским врачам и именно от их тактики во многих случаях зависит дальнейшая судьба пациентов.
Считаем необходимым напомнить, что еще в 60-е годы прошлого столетия сложными вопросами различных заболеваний нервной системы, в происхождении которых важную роль играет наследственная предрасположенность, занимался известный ученый с мировым именем С.Н. Давиденков, описавший лопаточно-перонеальную форму миопатий. В своих работах он впервые в Советском Союзе поднял проблему возможных прогнозов потомства при некоторых наследственных заболеваниях. С тех пор диагностика многих наследственных болезней поднялась на должную высоту благодаря новейшим молекулярно-генетическим технологиям. Однако способы определения типа наследования патологии остаются прежними, и каждому врачу важно помнить о том, что в случае доминантного типа «…риск передачи болезни потомству настолько велик, что больные члены семьи должны воздерживаться от деторождения. Наоборот, члены семьи, оставшиеся здоровыми, могут свободно иметь детей без всякой опасности передать им семейное заболевание» [4].
Методы диагностики
Несмотря на частое обнаружение генетической мутации во время генетического исследования, у некоторых больных с выраженной клинической картиной они отсутствуют. Из-за этого генетический анализ крови не является основным критерием заболевания. В большинстве случаев миопатию Ландузи-Дежерина диагностируют на основании характерных клинических проявлений, в первую очередь поражение типичных для болезни мышц.
Также дополнительно проводят следующие виды обследования:
- биохимический анализ крови с определением уровня креатинфосфокиназы;
- биопсию мышц;
- электронейромиографию.
Значительное повышение уровня креатинфосфокиназы свидетельствует о прогрессирующей атрофии мышечной ткани. У больных отмечается слабая активность миоцитов. Для гистологического исследования производят забор образцов тканей с разных участков тела. При исследовании обнаруживаются характерные признаки атрофии. Дополнительно больные нуждаются в офтальмологическом обследовании и консультации невропатолога.
Важно! Течение некоторых болезней схоже с данной миопатией. Врачи обязательно проводят дифференциальную диагностику с неврологическими заболеваниями, аутоиммунными и патологиями соединительной ткани.
Сопутствующие заболевания
Болезни мышц могут прогрессировать очень быстро, если их течение осложняют другие заболевания, уменьшающие подвижность. Поэтому мы обязательно лечим сопутствующие заболевания.
Основные заболевания, способные ухудшать самочувствие:
- Болезни суставов и позвоночника (артроз, грыжа межпозвонкового диска, остеохондроз и т.п.) блокируют подвижность спины и конечностей. Они могут развиться из-за смещения центров тяжести тела на фоне слабости мышц. Здесь мы с успехом применяем мягкую мануальную терапию, массаж, электрофорез карипазима.
- Хронические тонзиллит, фарингит, гайморит, аденоидит. Постоянное присутствие болезнетворных микробов в дыхательных путях обусловливает постоянную интоксикацию, а токсины негативно воздействуют на мышцы, особенно сердечную. Поэтому в случае необходимости мы выполняем курс лечения этих заболеваний.
- Другие хронические инфекции (почек, печени, нижних дыхательных путей).
- Избыточный вес. Он опасен избыточной нагрузкой на мышцы, суставы, позвоночник. Часто служит причиной обездвиженности. Мы выявляем причины ожирения и подбираем схему его лечения.
- Нарушения в работе щитовидной железы. Нарушения обмена веществ при снижении или повышении функции щитовидной железы могут быть причиной снижения силы мышц. При необходимости мы контролируем содержание щитовидных гормонов крови и привлекаем к лечению эндокринолога.
- Сосудистые болезни (артериальная гипертония, нарушения мозгового кровообращения) способствуют перегрузке сердца и быстрому развитию миопатии сердечной мышцы, а значит и сердечной недостаточности. При необходимости мы выполняем нагрузочные кардиопробы и подбираем лечение.
- Бронхиальная астма приводит к кислородному дефициту в мышечной ткани, что способствует слабости мышц и прогрессированию миопатии. При необходимости рекомендуем курс лечения по поводу бронхиальной астмы.
Лечение
Заболевание относится к неизлечимым диагнозам. Патогенетическое лечение отсутствует. Также достаточно сложно остановить быстропрогрессирующую атрофию. Лечение миопатии Ландузи-Дежерина направленно в первую очередь на замедление дистрофических изменений, приводящих к невозможности самостоятельно передвигаться и обслуживать себя, восстановление утраченных функций и общее улучшение состояния пациента. Медикаментозная терапия основана на применении медикаментов, поддерживающих миоциты, улучшающих обменные процессы в них, и благотворно воздействующих на сердце. Наиболее часто назначают такие препараты: АТФ-лонг, Милдронат, Метион, витамин Е, Цитофлавин, кокарбоксилаза.
Также для улучшения состояния значение имеют следующие факторы:
- питание – рекомендуется увеличить количество белков и сократить жиры;
- физиопроцедуры – больным показан массаж, электрофорез и другие процедуры;
- лечебная физкультура – умеренные физические нагрузки и специальный комплекс необходим для предупреждения развития сколиоза и контрактур.
Учитывая, что миопатия зачастую возникает в детском возрасте, особое внимание необходимо уделять психическому состоянию пациентов. Часто патология приводит к депрессивным расстройствам, нервным срывам и нарушениям эмоциональной сферой. Чтобы предотвратить психологические нарушения и улучшить эмоциональное состояние пациентов, им необходима помощь психолога или психотерапевта.
Для социализации необходимо помочь больным с выбором профессии, чтобы сохранить их работоспособность.
Посмотрите видео по теме статьи!
Возможные осложнения
Несмотря на тщательное изучение, миопатия Ландузи-Дежерина продолжает относиться к тяжелым патологиям. Во многом прогноз зависит от особенностей течения и скорости развития патологического процесса. Гипомимия и амимия не только ограничивает физические возможности, но и является причиной психологического дискомфорта и трудностей с социализацией.
Прогрессирование и вовлечение в патологический процесс других частей тела приводит к значительным ограничениям, вплоть до невозможности передвигаться.
В некоторых случаях при поражении круговых мышц глаза и частых воспалительных процессах у пациентов ухудшается зрение вплоть до слепоты.
Зачастую, выполнение рекомендаций врача помогает сохранить продолжительность жизни, но при крайне тяжелой форме заболевания возможен летальный исход из-за тяжелой дыхательной недостаточности.
Массаж при миопатии и амиотрофии
Стандартные методики массажа не вполне подходят для лечения миопатии и амиотрофии. Кроме того, эффект массажа напрямую зависит от правильности его выполнения. К сожалению, нам регулярно приходится видеть пациентов после курса массажа с абсолютно непроработанными при массаже проблемными мышцами.
Правильный массаж начинается с определения неэластичных, уплотненных, ослабленных участков мышц. Именно на эти участки и должны быть направлены основные усилия при массаже. Причем требуется сочетание тонизирующего массажа на ослабленные участки с расслабляющим, растягивающим и рассасывающим на уплотненные участки мышц.
Если имеется слабость дыхательной мускулатуры, выполняется массаж грудной клетки для облегчения дыхательных движений.
Мы выполняем массаж в клинике и обучаем родственников пациентов самостоятельному выполнению массажа в домашних условиях. Если пациент находится на лечении без сопровождающих, обучаем самомассажу.
Что нужно запомнить?
- Миопатия Ландузи-Дежерина характеризируется прогрессирующей атрофией мышц лица, плеч и лопаток.
- Основной причиной заболевания является генетическая предрасположенность.
- Признаками патологии является гипомимичность и амимичность, мышечная слабость и потеря мышечной ткани.
- Лечение направлено на торможение развития болезни и предотвращение ее прогресса.
- Мышечная дистрофия приводит к инвалидизации пациента из-за выраженных ограничений его возможностей.
- Прогноз на выздоровление неблагоприятный, так как болезнь неизлечима.
Литература
- Асанов А. Ю. Основы генетики и наследственные нарушения развития у детей: Учеб. пособие для студентов вузов. — М.: Академия, 2003. — С. 216.
- Бадалян Л. А. Детская неврология: учеб. пособие. 4-е изд. — М.: МЕДпресс информ, 2021. — 608 с.
- Влодавец Д. В., Сухоруков В. С., Харламов Д. А., Белоусова Е. Д. Способ лечения врожденных структурных миопатий и врожденных мышечных дистрофий путем коррекции вторичных митохондриальных изменений: Автореф. дис. … к. м. н. — М., 2009. — 28 с.
- Гринио Л. П. Атлас нервно-мышечных болезней. — М.: АНС, 2004. —167 с.
- Ерохина В. А. Реабилитация детей с наследственными миопатиями // Вестник Московского Университета МВД России. — 2015. — № 12. — С. 304—308.
- Arvanitidis A., Henriksen K., Karsdal M. A., Nedergaard A. Neo-epitope Peptides as Biomarkers of Disease Progression for Muscular Dystrophies and Other Myopathies // J Neuromuscul Dis. — 2016. — № 30. — Р. 333—346.
- Carroll M. B., Newkirk M. R., Sumner N. S. Necrotizing Autoimmune Myopathy: A Unique Subset of Idiopathic Inflammatory Myopathy // J Clin Rheumatol. — 2021. — № 22. — Р. 376—80.
- Inoue M., Nishino I. Diagnosis of Idiopathic Inflammatory Myopathy: A Muscle Pathology Perspective // Brain Nerve. — 2021. — № 68. — Р. 1431—1441.
Диагноз – миопатия (амиотрофия). Что делать?
Сегодня панацеи от наследственных мышечных дистрофий не существует. Однако правильное лечение позволяет ощутимо затормозить атрофию мышц и увеличить регенерацию и рост новой мышечной ткани, и даже вернуть некоторые утраченные возможности. Лечение миопатии и амиотрофии требует повседневного выполнения ряда медицинских процедур, поэтому мы не только оказываем лечебную помощь, но и обучаем родственников пациентов и/или самих пациентов самостоятельному выполнению необходимых процедур.
Лечение в нашей клинике включает в себя следующее:
- Прием лекарств по схеме, которая расписывается на срок 3-12 месяцев;
- Лечебное питание;
- Физиотерапия;
- Массаж;
- Гимнастика;
- Психотерапия и духовные практики;
- Нейропсихологическое развитие (для детей, отстающих в интеллектуальном развитии).
По каждому из перечисленных пунктов проводится подробное обучение. Мы настаиваем на обучении самостоятельному выполнению процедур, т.к. стремимся сделать регулярное и достаточное по объему лечение еще и доступным и дешевым.