Progressive paralysis (paralytic dementia, Bayle's disease) is an organic pathology marked by progressive disturbances in psychological and neurological activity, up to the onset of dementia. All processes can be accompanied by somatic and neurological disorders.
The likelihood of a patient developing progressive paralysis varies from 1 to 5% of all people who have had syphilis. Among other things, the male part of patients suffers from the disease more often - according to statistics, 5 times than women. The incubation period of the pathology itself is 10-15 years from the moment of infection.
The cause of the development of this disease is syphilis. However, the risks may increase with the development of other sexually transmitted diseases, such as gonorrhea.
Stages of the pathology
In its course, Bayle's disease goes through 3 stages, namely:
- Initial – its duration varies from 2 months to six months. At this stage, patients will complain of loss of strength and poor sleep, lethargy and dementia, they manifest themselves with attacks of sentimentality and excessive sleepiness during the day and insomnia at night. In the process of examining and diagnosing this stage, doctors check the blood reaction according to the Wasserman method, which, if pathology is present, will be positive. A reaction is also carried out according to Lanta, and the level of protein in the blood is increased.
- Full development stage . At this stage, patients behave inappropriately, lose their sense of tact, giving way to a feeling of euphoria and complacency, and develop inherently delusional ideas, groundless joy or tears. The patient experiences disorders that affect speech and handwriting, some may rapidly lose weight, others may, on the contrary, develop one or another degree of obesity, the face swells, and the skin turgor changes. Abscesses and damage to the body by boils, excessive fragility of bones, and degenerative lesions of the heart and liver can develop negatively. The course of this stage is diagnosed using the Wasserman blood reaction - the results will be positive, like other reactions of serological studies.
- Dementia - at this stage of the disease, the patient has a decrease in self-criticism and a weakening of his own judgments, conclusions are absurd and delusional, sometimes replaced by a feeling and state of euphoria. As the pathology progresses, the patient will lose interest in the world around him, will not respond to external stimuli and answer questions, and urinary and fecal incontinence will develop. At this stage, the patient’s limbs may become covered with ulcers, strokes may lead to death, bone fragility increases significantly, and atypical paralysis, progressive in nature, may also develop.
Medical Internet conferences
Introduction. Progressive supranuclear palsy (PSP), or Steele-Richardson-Olszewski disease, is a degenerative disease of the brain. PSP is characterized by progression of akinetic-rigid syndrome, early development of postural instability, combined with supranuclear ophthalmoplegia, pseudobulbar syndrome and frontal-type dementia. The disease was first described in 1963-1964. Canadian neurologists J. Steel and J. Richardson together with pathologist J. Olszewski. The authors presented 7 cases of an unknown neurodegenerative disease presenting with neck and upper trunk stiffness, pseudobulbar abnormalities, supranuclear ophthalmoplegia, and dementia. In our country, PSP was first described in 1980 on the example of two patients observed in the clinic of nervous diseases of the Moscow Medical Academy. THEM. Sechenov. According to various population studies, PSP is detected in 4-7% of cases of parkinsonism, its prevalence reaches 5 × 10-5. The onset of the disease occurs in middle and old age; men are affected somewhat more often.
The etiology and pathogenesis of PSP have not been fully studied. The disease belongs to the group of “tau-pathies”. This group, in addition to PSP, includes Alzheimer's disease, Pick's disease, parkinsonism-ALS-dementia, corticobasal degeneration, as well as a number of hereditary diseases (hereditary multisystem tauopathies).
Morphologically, PSP is characterized by degenerative changes in neurons with the formation of neurofibrillary tangles in the substance of the midbrain, pons, basal ganglia and dentate nuclei of the cerebellum. Neurochemical changes are characterized by a decrease in the concentration of dopamine and its metabolites in the caudate nucleus and putamen.
The vast majority of cases of PSP are sporadic, but De Yebenes et al. in 1995, and then other authors, described families with a presumably autosomal dominant pattern of inheritance of the disease. According to molecular genetic studies, one form of PNP (PSNP1) can be caused by a heterozygous mutation in the gene, which is localized on 17q21.31 and encodes tau protein.
The disease begins with such nonspecific symptoms as increased fatigue, depressed mood, dizziness, headache, arthralgia, drowsiness or insomnia, decreased performance, narrowing of the sphere of interests and circle of friends. Over time, more specific symptoms come to the fore: postural instability with frequent falls (60% of cases), dysarthria (33%), slowness of movements (13%), visual disturbances (13%). Very rarely, initial symptoms include dysphagia, freezing when walking, behavioral changes, or vertical gaze palsy.
At the onset of the disease, as a result of damage to the nigrostriatal segment, characteristic muscle rigidity and progressive oligobradykinesia develop. Parkinsonism in PSP is symmetrical, develops early, and is more pronounced in the axial muscles than in the limbs; a typically characteristic increase in tone in the extensors of the neck and back (“proud posture”). Rest tremor is usually absent, and most often parkinsonism syndrome is represented by an akinetic-rigid form that is not amenable to levodopa therapy. The gait with PSP can differ significantly from that with Parkinson's disease and has the character of “parkinsonian ataxia”: the patient cannot correctly coordinate the movements of the torso and legs in relation to the center of gravity, which leads to frequent falls back without attempts to maintain balance.
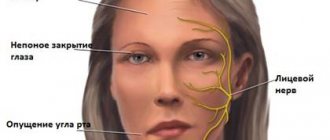
Later, supranuclear oculomotor disorders appear, in the pathogenesis of which degeneration of the dorsal parts of the midbrain plays a decisive role. Eye movements are disrupted first in the vertical plane, and subsequently complete ophthalmoplegia gradually develops with retraction of the upper eyelids and the appearance of a characteristic “surprised” facial expression.
Relatively early, patients with PSP also develop severe pseudobulbar manifestations - dysarthria, dysphagia, reflexes of oral automatism, forced laughter and crying.
A significant proportion of patients, already at an advanced stage of the disease, experience cognitive and emotional-personal disorders reflecting dysfunction of the frontal lobes.
When making a diagnosis, the entire clinical picture as a whole, as well as the features of its development, must be taken into account. The NINDS-SPSP criteria, which have high specificity, are widely used to make a diagnosis [1-7].
Important problems in the management of patients with PSP are the difficulty of diagnosis, especially in the early stages of the disease, as well as the low effectiveness of treatment with antiparkinsonian drugs.
Target. Presentation of our own observation of PSP with atypical onset and course.
Patient P., 54 years old, was admitted to the clinic with complaints of limited movement of the eyeballs; stiffness, awkwardness and trembling in the limbs, more in the left; gait disturbance, drooling, urinary incontinence.
From the anamnesis it is known that she has been ill since the age of 49, when restrictions on the movements of the eyeballs appeared, more on the left, symmetrical drooping of the eyelids, and general weakness.
A neurological examination revealed bilateral ophthalmoparesis: symmetrical hemiptosis of the upper eyelids, paresis of downward and upward gaze, limited mobility of the eyeballs to the sides. There were no motor disorders in the limbs, muscle strength was sufficient, reflexes were preserved and symmetrical. Based on the results of the examinations (needle EMG, histological examination of muscle biopsy), information about the previously diagnosed progressive muscular dystrophy (PMD) in the patient’s mother, which manifested itself at the age of 60 years with oculomotor disorders, it was assumed that the patient had ophthalmoplegic Graefe PMD.
Three years later, stiffness and awkwardness appeared in the left limbs, and then a slight tremor in them. Within a year, similar symptoms appeared in the right extremities. Since that time, she began to notice tension in the neck muscles, gait disturbance, drooling, and occasional urinary incontinence.
When examined at the age of 54 years, the leading neurological status was extrapyramidal syndrome: oligobradykinesia more pronounced in the axial parts, rapidly increasing slowness of movements of the head and torso; gross hypomimia, giving the face a mask-like appearance; muscle rigidity with a predominance in the extensor muscles of the neck and back, the “cogwheel” phenomenon; low-amplitude resting tremor in combination with moderate postural-kinetic trembling hyperkinesis in the hands, with a predominance on the left. When walking, subcortical astasia was detected, the arms were bent at the elbows. There were severe oculomotor disturbances to the degree of ophthalmoplegia on both sides. Reflexes from the limbs are high and symmetrical. Significant autonomic failure (hypersalivation, periodic urinary incontinence).
Neuropsychological examination revealed moderate cognitive impairment (MMSE: 25 points, MoCA test: 23 points).
MRI of the brain revealed atrophy of the frontal and temporal lobes, expansion of the third and fourth ventricles, and atrophy of the midbrain tegmentum.
Repeated needle EMG and ENMG revealed no signs of primary muscle lesions or disorders of neuromuscular transmission.
The information obtained allowed us to change the diagnosis: Progressive supranuclear palsy (Steele-Richardson-Olszewski disease); severe parkinsonism syndrome, akinetic-rigid-tremor form; ophthalmoplegia; postural instability; mild cognitive impairment; autonomic failure (hypersalivation, occasional urinary incontinence).
During an observation period of more than 1 year, attempts were made to select antiparkinsonian drugs; the patient was prescribed combinations of antiparkinsonian drugs in maximum daily doses using various groups (dopamine receptor agonists, amantadines, levodopa drugs), but no significant clinical effect was achieved.
Conclusion. A feature of this clinical case is the atypical onset of PSP with the development of ophthalmoparesis. The subsequent development of parkinsonism and postural disturbances is typical of the clinical picture of PSP. However, the severe autonomic failure that was detected in the patient is not typical for this disease.
Clinical forms of progressive paralysis
Each of the presented forms of the disease is marked by its own special, clinical form of the pathology, and it is on this issue that it is worth dwelling in more detail:
- Simple form - at this stage, the pathology is marked by a characteristic increase in such a symptom as dementia, accompanied by a certain carelessness, inappropriate actions, loss of a sense of tact, indifference to the environment and situation, a decrease in the ability to remember, and a weakening of memory is observed.
- Expansive - this form is characterized by the fact that the patient has an elevated mood and ideas that are absurd in their manifestation, verbosity and delusions of grandeur.
- Depressive-hypochondriacal manifestation of pathology - this stage is marked by depression and bouts of tearfulness, complaints of a hypochondriac and Cotard's syndrome.
- Agitated manifestation of the syndrome - in this case, there is pronounced motor excitability and the patient’s tendency to destruction, manifestations of attacks of aggression. But the most important thing is hallucinations that affect hearing and vision.
- Circular manifestation of pathology - when diagnosing this form of the disease, the patient exhibits manic attacks and depression, alternating attacks of lethargy and euphoria, inactivity, which is replaced by a gloomy and gloomy mood, which is replaced by a feeling of dysphoria.
- Hallucinatory-paranoid manifestation of pathology - at this stage, true manifestations of hallucination are noted, little and insignificantly systematized, quite a lot of delusional ideas in such a relationship as idea and influence, catatonic disorder itself.
- The catatonic type of the pathological course of the disease is marked in its manifestation with the so-called catatonic stupor, a certain degree of inhibition or excitement. They are, according to some psychologists, very unfavorable signs.
- Galloping and slow-onset forms of the disease . If we talk about the galloping form, it is marked by a very rapid course, when the patient expresses active motor agitation, delusions and epileptic seizures, vegetative, as well as trophic disorders, severe exhaustion and, in some cases, death. The pathological stage can last from 1-2 weeks to several months. If we talk about a slowly ongoing stage, we are talking about an atypical form of progressive paralysis. This pathological change is very rare and manifests itself as aphasic disorders and manifestations of apraxia.
- Senile paralysis - this symptom and form of pathology is most often diagnosed in patients aged 60 years or more and is characterized by a very long course, about 35-40 years, and as a result, death. This phase is very similar to the manifestations of senile psychosis.
- Infantile and juvenile paralysis is most often diagnosed in children and adolescents in the age group from 6 to 16 years. The reason for the development of this form of pathology is infection of the fetus through the placenta of the fetus from the mother with syphilis - the so-called congenital pathology. Most often, the disease manifests itself as a delay in the mental development of the child, as well as damage to the inner ear and deformation of the front teeth, and epileptic seizures.
- Taboparalysis – in this case we are talking about a combination of a progressive form of paralysis and tabes dorsalis. Symptoms of this pathology manifest themselves in the form of absence of the knee and Achilles reflexes, loss of sensitivity, depression and nihilistic delirium are somewhat less common. Symptoms can manifest themselves as a sharp loss of weight or a sharp gain in weight, the appearance of puffiness on the face, changes in facial turgor, the appearance of abscesses and boils, and excessive bone fragility.
Anatomical data
With progressive paralysis, nerve cells in the brain and often the spinal cord die. The arrangement of ganglion cells in rows and layers is disrupted. Glia cells and fibers grow, increase in number and thicken. Already with a superficial examination, it is possible, based on the degree of increase in the number and especially thickening of these elements, to distinguish this proliferation of glia from that which occurs in senile forms. Mitoses are often observed in glial cells. The membranes of small vessels of the cerebral cortex (and other organs) are infiltrated by round cells, mostly plasmatic in nature. The latter are considered characteristic of progressive paralysis, since they are not observed in any other disease except sleepy, which does not occur in our country. New capillary formation can often be clearly seen.
Macroscopically, there is a decrease in the brain; in advanced cases, the weight reaches 1000 grams, the surface is uneven due to the process of wrinkling, the gyri are narrowed, the furrows are widened. The white matter is dirty in color and contracts when cut, unless the atrophy is masked by cerebral edema.
Nowhere is hemorrhagic pachymeningitis as common as in progressive paralysis; the dura mater is often fused to the skull.
Secondary degenerations are also observed in the spinal cord, since there are brain lesions, but in addition, the primary changes that occur in it are often the same as in the brain.
Chronic degeneration is sometimes observed in the peripheral nervous system. The aorta is mostly syphilitically changed. And other organs, especially the liver, do not remain normal. It should be noted that the usual manifestations of syphilis and their consequences (with the exception of vascular phenomena) are rare in paralytics.
Symptoms of pathology
Symptoms of progressive paralysis will manifest themselves as follows:
- speech impairment and failure in the ability to connect complex words;
- a patient diagnosed with progressive paralysis loses the ability to do basic math, and his motivation decreases significantly;
- the patient's consciousness is clouded;
- memory is lost - both short-term and long-term;
- muscle atrophy and weakness are observed in the upper and lower extremities, as well as weakness in other parts of the body;
- attacks of delirium and hallucinations, increased attacks of irritability and irascibility, which are followed by attacks of depression and moodiness;
- muscle cramp.