- How common are neuroblastomas?
- Risk factors for neuroblastoma
- What are the causes of neuroblastoma?
- Is it possible to prevent the development of neuroblastoma?
- Is early diagnosis of neuroblastoma possible?
- How is neuroblastoma diagnosed?
- Treatment of neuroblastoma in children
- What happens after treatment for neuroblastoma ends?
Neuroblastoma is a type of malignant tumor and usually occurs in infants and children and very rarely in children over 10 years of age. The cells of this tumor resemble nerve cells in the early stages of their development in the fetus.
One third of neuroblastomas develop in the adrenal glands, the other third in the abdominal cavity along the nerve trunks along the spine, and the rest in the chest cavity and neck. Some neuroblastomas arise from the spinal cord. Sometimes, due to the wide distribution of the tumor at the time of diagnosis, it is difficult to determine the exact location where the tumor originated.
Not all tumors of the nervous system are malignant.
Ganglioneuroma is an example of a benign tumor.
Ganglioneuroblastoma is a mixed tumor and contains both malignant and benign areas. Immature (malignant) cells from this tumor can grow and metastasize.
Ganglioneuromas are usually removed surgically and then carefully examined under a microscope to determine whether there are areas of ganglioneuroblastoma. If ganglioneuroma is diagnosed, no additional treatment is required. In contrast, ganglioneuroblastomas are treated in the same way as neuroblastomas.
Neuroblastoma is an unusual tumor for many reasons. This tumor secretes a hormone that leads to unusual changes in the body, for example, rotational movements of the eyeballs, spastic muscle twitching, and the presence of constant loose stools.
These changes are called paraneoplastic syndromes.
Neuroblastoma can behave in unusual ways. Sometimes tumor cells spontaneously die and the tumor disappears. This phenomenon occurs most often in very young children and much less frequently in older patients. In some cases, tumor cells spontaneously mature and stop dividing. Thus, neuroblastoma turns into ganglioneuroma, a benign tumor.
How common are neuroblastomas?
Neuroblastoma is the most common tumor in infants and is the fourth most common malignant tumor in older children, after acute leukemia, central nervous system tumors, and malignant lymphomas.
Every year, 6-8 children per 1 million children under 15 years of age (average age 2 years) become ill with neuroblastoma in Russia.
Approximately 650 new cases of neuroblastoma are diagnosed each year in the United States, a number that has remained stable for many years. Neuroblastoma occurs slightly more often in boys than in girls. For every 6 cases of tumors in boys, there are 5 cases in girls.
The average age of patients at the time of diagnosis is 17 months. One third of cases are diagnosed in children under 1 year of age. Almost 90% of neuroblastomas are detected before 5 years of age. Only 2% of tumors are detected after the age of 10 years and in adult patients. In rare cases, neuroblastoma can be detected by ultrasound before the baby is born.
In 7 out of 10 cases, neuroblastoma already has metastases at the time of diagnosis.
Chapter 10.
Neuroblastoma (the currently generally accepted name for malignant tumors of the sympathetic nervous system) is the most mysterious tumor of childhood, both from a clinical and biological point of view. In the structure of all cancer incidence, neuroblastoma accounts for 7-11% of the total number of malignant tumors in children, ranking fourth after acute leukemia, central nervous system tumors and malignant lymphomas. The incidence of neuroblastoma is 0.85-1.1 per 100,000 children under 15 years of age. The age distribution of neuroblastoma throughout childhood is heterogeneous and the frequency of diagnosis decreases as the child grows older. In children of the first year of life, neuroblastoma is the most common malignant tumor; its incidence at this age is 6.1 per 100,000 children under one year of age. The incidence of neuroblastoma at the age of 1 to 5 years is 1.7 per 100,000 children, and at the age of 5 to 10 years it is 0.2 per 100,000 children of this age. The name “neuroblastoma” was proposed by J. Wright, who in 1910 showed that a number of tumors of the retroperitoneum and posterior mediastinum have a clear morphological similarity to the tissue of the developing sympathetic nervous system. The source of tumor growth in neuroblastoma is elements of the sympathetic nervous system, so theoretically neuroblastoma can arise almost anywhere in the body where sympathetic ganglia or paraganglia are present.
Neuroblastoma is a typical representative of embryonal tumors, so it is mainly diagnosed in young children (the average age at the time of diagnosis is 2 years). 90% of cases are diagnosed before 5 years of age. However, this tumor can also occur in adolescents and even adults, although it is extremely rare. The predominance of tumors of the sympathetic nervous system in children is due to the peculiarities of the development of this system in humans, since the formation of sympathetic ganglia does not end in the prenatal period and their cellular structure becomes the same as in adults (Fig. 10-1), only by the age of 5 years of a child’s life. Naturally, with high intensity and intensity of the processes of growth and development of the sympathetic nervous system, conditions are created for the occurrence of tumor proliferation.
According to a number of authors, the frequency of neuroblastoma “in situ”, which is found in the adrenal glands of children under 3 months of age who died from other causes, is approximately 1:200. Since far fewer children subsequently develop classic neuroblastoma, the potential for spontaneous regression of neuroblastoma “in situ” should be very high. Although it must be kept in mind that since neuroblastic nodules may be a normal part of embryonic development, a clear division between true neuroblastoma in situ and primitive neuroblastic neural crest cells may not be possible in some cases. The cause of neuroblastoma is unknown, although it is still certain that the malignant process is associated with changes in the DNA of cells. These changes and their impact on the forecast are reflected in Table 10-1:
It is well known that characteristics associated with a favorable prognosis are detected at stages 1, 2 and 4S of the process. Whereas DNA euploidy, deletion of the short arm of chromosome 1 (del 1p 36, 2-3) and N-myc amplification are recorded in common and unfavorable stages (3 and 4). In this regard, today the question is being seriously discussed whether the prognostically favorable stages of neuroblastoma (1,2 and 4S) and prognostically unfavorable (3 and 4) stages are different biological types of the disease.
Biology
Neuroblastoma belongs to a group of embryonal tumors such as hepatoblastoma, nephroblastoma, and embryonal rhabdomyosarcoma. All of them are characterized by manifestation at an early age and have similar cytomorphological characteristics characteristic of embryonal tumors. Neuroblastoma is distinguished by a number of specific, unique features of its biological behavior that are not characteristic of other malignant tumors.
1. The ability for spontaneous regression. In clinical practice, there are well-known examples where in infants with the classic picture of stage 4S neuroblastoma (usually with massive liver damage), involution of the malignant process is observed from about 4 months of age. Only a very small proportion of these patients require minimal chemotherapy or radiation therapy to “induce” the regression process. So far, no marker is known that determines the fracture during the course of the disease from progression to regression. It is also unknown why a number of patients under 1 year of age with a seemingly 1U-S stage of the process do not experience tumor regression. The factors causing this process in groups of patients with neuroblastoma who do not belong to the stage 4S category have also not been determined.
2. Ability to differentiate (“maturation”). Another surprising property of neuroblastoma tumor cells was noticed during a cultural study: a culture of cells taken from an aggressively growing tumor acquired the features of differentiating nervous tissue during the cultivation process. Various agents are capable of inducing this process in vitro: retinoic acid, the so-called nerve tissue growth factor, some cytostatics, papaverine. However, to date, there have been no reports of successful induction of the differentiation process in vivo. The first clinical observation of the “maturation” of sympathogonioma into benign ganglioneuroma 10 years after diagnosis and without any treatment was described back in 1927. According to German authors (F. Berthold), differentiation of malignant neuroblastomas into benign tumors occurs quite rarely (1:1150), although convincingly.
3. The ability for rapid aggressive development and rapid metastasis. The main factors related to undoubted markers of progression are N-myc amplification and deletion of 1 pair of chromosome (1p). The clinical variety of neuroblastomas is accompanied by certain genetic abnormalities: quantitative changes in DNA, amplification of the N-myc proto-oncogene, and deletion of the short arm of chromosome 1. Generally speaking, the number of chromosomes (DNA content) divides these tumors into 2 groups. Diploid tumors are more common in older children, while triploid tumors are predominantly registered in children under 1 year of age.
Clinical picture
The clinical picture of the disease depends on the location of the primary tumor, the presence and location of metastases, and the amount of vasoactive substances produced by the tumor. The main complaints are pain (30-35%), fever (25-30%), weight loss (20%). The presence of complaints and their number mainly depends on the stage of the disease. Thus, according to German colleagues, the asymptomatic course of neuroblastoma was registered in patients with stage 1 in 48% of cases, with stage 2 in 29%, with stage 3 in 16%, with stage 4 in only 5%, with stage 4 - in in 10% of patients. Damage to the cervicothoracic section of the sympathetic trunk early causes Horner's syndrome, which makes possible relatively early diagnosis of tumors arising from these sections. Localization of the tumor in the posterior mediastinum can cause an obsessive cough, respiratory disorders, deformation of the chest wall, can cause dysphagia, and in babies - frequent regurgitation. Damage to the bone marrow leads to anemia and hemorrhagic syndrome. A characteristic symptom of “spectacles” with exophthalmos is when the retrobulbar space is affected in children with stage 4 of the disease. Metastases to the skin have a bluish-purple color and a dense consistency. When the process is localized in the retroperitoneal space, palpation reveals a lumpy, rocky density, practically non-displaceable tumor (early fixation of the tumor occurs due to rapid growth into the spinal canal through the intervertebral foramina). The spread of the tumor from the chest cavity into the retroperitoneal space through the diaphragmatic openings causes the same “hourglass” or “dumbbell” symptom.
In principle, neuroblastoma can arise in any organ that has sympathetic innervation, but typical sources of tumor growth in neuroblastoma are the sympathetic nerve trunk along its entire length and the adrenal medulla (Fig. 10-2). Approximately 40% of tumors arise in the adrenal glands, 30% originate from the lumbar sympathetic trunk, 15% from the thoracic region, 3% from the pelvic paraganglia, and 1% from the cervical region.
According to various authors, the share of atypical localizations or neuroblastomas with an unidentified primary focus is from 5 to 15% (Fig. 10-3). The most characteristic clinical picture is with the retroperitoneal (most common) tumor localization: a dense, practically non-displaceable tumor. General symptoms are very common: weight loss, weakness, bone and joint pain, anemia, fever. Quite often, a child is examined for a long time for bone pain with diagnoses: arthritis, epiphysitis - rheumatoid or nonspecific, until general symptoms of the disease develop.
An increase in blood pressure may be associated either with excessive secretion of catecholamines by the tumor or with compression of the renal vessels. Intractable diarrhea, a symptom, although rare, can mask the true cause of intestinal disorders for a long time.
Neurological symptoms can be expressed in children whose tumor is localized paravertebrally with penetration into the spinal canal, or with primary intraspinal localization.
A specific group of patients with cerebellar ataxia, which is usually associated with the localization of a primary well-differentiated tumor in the mediastinum, has a good prognosis. The mechanism of toxic effects on the cerebellum is not clear. It is assumed to be of immunological origin due to cross-reactivity of autoantibodies between neuroblastoma and cerebellar nerve tissue. The nystagmus that accompanies this condition gives the name “dancing eyes” syndrome.
Unfortunately, more than half of the patients at the time of diagnosis have stage 4 of the process, i.e. they have distant metastases (Table 10-3)
The lungs are the organ most often affected in stage 4 of any malignant tumor; in neuroblastoma they are affected extremely rarely.
Diagnostics.
According to international criteria, the diagnosis of neuroblastoma can be established by histological examination of biopsy material obtained from the primary tumor (or from metastases), or by identifying bone marrow lesions in combination with an increased content of catecholamines or their derivatives: vanillylmandyl (VMC), homovanillic (HVA) acids and dopamine in the blood or urine. Levels of VMC and HVA are elevated in 85% of patients and dopamine levels in 90% of patients with neuroblastoma. The level of excretion of catecholamines does not affect the prognosis, while a high ratio of HVA: VCH (the relationship is directly proportional) indicates the presence of a poorly differentiated tumor and is associated with a worse prognosis.
Page 191
Tumor markers.
Neuroblastoma belongs to the category of childhood tumors in which tumor markers are used for diagnosis, monitoring during treatment, and as prognostic factors.
The metabolites of catecholamines are highly specific and easily determined: VMC, GVA and Dopamine (DA). The level of these substances is determined in the child’s daily urine and blood serum. Diagnostically significant is an increase in the content of VMC, GVA and DA by 3 times in comparison with the age norm. Interestingly, the method for determining catecholamine derivatives in urine is approximately 15% more sensitive than in serum. In the case of false-negative results for the determination of catecholamine metabolites (which occurs in approximately 15% of patients), the determination of the serum level of neuron-specific enolase (NSE), an enzyme detected in neurons, will help in diagnosis. In 1981, K. Tapia showed that tumors of neurogenic origin have a high content of NSE. An increased level of this enzyme in the blood serum is characteristic not only of neuroblastoma, but also of other tumors of neuroectodermal origin (PNET, Ewing's sarcoma), Wilms tumor, and some types of leukemia. The level of NSE content in serum also has prognostic significance, so an NSE value of more than 80-100 ng/ml indicates an extremely unfavorable prognosis. Another biochemical marker of neuroblastoma is ferritin. In a number of patients with neuroblastoma, ferritin levels are elevated, and its amount in the serum is directly proportional to the volume of the tumor mass. It is known that ferritin has some biological effects that negatively affect the patient’s immunity and therefore increased ferritin levels are also associated with a worse prognosis in patients with neuroblastoma.
The enzyme lactate dehydrogenase (LDH) does not belong to the markers specific for neuroblastoma, but its content in the serum has prognostic significance for these patients: its elevated level is more often observed in advanced stages of the disease, which probably explains the connection between elevated LDH levels and an unfavorable prognosis . Perhaps the LDH level only reflects the rate of tumor proliferation and therefore, in common processes with an obviously unfavorable prognosis, a high LDH level is recorded.
Other markers of neuroblastoma are Ganglioside GD2, neuropeptide Y and Chromogranin A. The detection of these markers by immunohistochemistry serves as a criterion for the diagnosis of neuroblastoma, and their effect on the prognosis of the disease is being studied.
Scheme for determining the degree of spread of the tumor process.
1. Location of the primary tumor - ultrasound, CT, MRI. 2. Chest - radiography, CT. 3. Abdominal cavity - ultrasound, CT. 4. Osteoscintigraphy with Te 99 and subsequent radiography of identified foci of hyperfixation of the isotope. 5. Scintigraphy with 131 J metaiodobenzylguanidine (MJBG)*. 6. Bone marrow aspiration biopsy (from 4-8 places). 7. Trephine biopsy of the bone marrow with histological and immunohistochemical studies. 8. Biopsy of lesions suspicious for tumor metastases.
X-ray and CT scans reveal characteristic calcifications in the tumor tissue (which is a good prognostic sign) and the number of which increases with a positive tumor response to chemotherapy (Fig. 10-5).
Histology
There are several histological classifications of the morphological structure of neuroblastoma (Shimada, Huge, Joshi), which are based on the degree of tumor differentiation: from highly differentiated and less malignant ganglioneuroblastoma to undifferentiated small round cell highly malignant neuroblastoma.
In typical cases, the tumor consists of small, uniform, round cells with a hyperchromatic nucleus surrounded by a minimal amount of cytoplasm. Neuroblastoma is characterized by the formation of pseudo-rosettes, called Homer-Wright rosettes: this is a peculiar morphological structure formed along the periphery by neuroblast nuclei with eosinophilic fibrils located in the center. The tumor often contains large areas of necrosis. Interstitial hemorrhage is relatively common in less differentiated tumors. Diffuse infiltration of lymphocytes is characteristic of more differentiated tumors. The presence of calcifications is a characteristic feature of neuroblastoma, and their number may increase as the tumor responds well to therapy.
Neuroblastoma belongs to the family of small round cell tumors and with simple light microscopy it is often very difficult for a morphologist to identify the nature of the tumor, especially since in different sections of the tumor the histological picture can differ significantly in cellularity, signs of maturation and differentiation, and in the amount of stroma. To accurately verify the diagnosis, an immunohistochemical study is almost always necessary.
To verify the diagnosis, cytogenetic research will also provide undeniable help: 70% of neuroblastomas have a deletion or rearrangement of the short arm of chromosome 1, which clearly correlates with a poor prognosis. Staging.
The outcome of the disease is determined mainly by the stage of the disease. For many years, clinicians used the Evans clinical classification, which has now been replaced by the International Classification (INSS), adopted in 1993 and which is a modification of the Evans classification.
According to various clinics, the distribution of stages in diagnosed patients with neuroblastoma: stage 1 -1-4%, stage 2 -10-15%, stage 3 - 20-25%, stage 4 - 50-60%. In children under one year old, the distribution of stages: stage 1 - 20-25%, stage 2 - 14-15%, stage 3 -20-25%, stage 4 -15-20%, stage 4 - 20-25%. In other words, the vast majority of pediatric oncologists deal mainly with patients who have a common process. Treatment.
When treating neuroblastoma, all three methods of antitumor treatment are used: chemotherapy, radiation therapy and surgery.
In the treatment of patients with localized stages 1 and 2A, radical tumor removal is most often sufficient. The presence of a microscopic residual tumor, according to many authors, almost never leads to relapse or metastasis, which distinguishes neuroblastoma from most other solid tumors. It is extremely important to biopsy regional lymph nodes on both sides of the tumor to accurately determine the stage of the disease. In patients with stage 2B, in addition to tumor removal, it is necessary to use chemotherapy and irradiation of the lesion and involved lymph nodes. Patients with stage 3 disease already have an unresectable tumor at the onset, so preoperative chemotherapy is necessary, which leads to a significant reduction in the size of the tumor, sometimes even until its radical removal is possible. In case of incomplete removal, radiation therapy to the site of the removed tumor can help cope with the residual tumor. Modern chemotherapy programs and improved surgical techniques (microsurgery) make it possible to cure up to 60% of patients with stage 3 neuroblastoma.
Patients with stage 4 disease, who make up the majority of patients, have a worse prognosis; 5-year survival rate, even with modern chemotherapy programs, is no more than 20%. The localization of metastases has a prognostic role for patients with stage 4 neuroblastoma: children who have metastases only to distant lymph nodes have a much better prognosis than patients with bone metastases. In the treatment of this group of patients with an extremely unfavorable prognosis, even the use of megadose chemotherapy (melphalan, Vepeside) and total body irradiation with autologous bone marrow transplantation did not bring the expected results. Research is currently underway on the use of a new class of chemotherapy drugs (topoisomerase 1 inhibitors, such as topotecan and irinotecan) and immunotherapy in patients with resistant forms of neuroblastoma and as the disease progresses. (St. Jude Children's Hospital, USA)
Modern standard chemotherapy regimens use platinum drugs (cisplatin, carboplatin), epipodophyllotoxins (VP-16, VM-26), dacarbazine, adriblastine, cyclophosphamide, ifosfamide, vincalcaloids (vincristine, vindesine).
According to various authors, the survival rate of patients with neuroblastoma in general is about 50% (49-55%), by stage: stage 1 - 100%, stage 2 - 94%, stage 3 60% (67-57%), stage 4 - 10-20%. Stage 4S - 75%.
Stage 4S neuroblastoma
This situation occurs almost exclusively in children under 1 year of age. Despite the presence of distant metastases in the skin, liver and bone marrow at this age, spontaneous regressions are very often observed a few months from the moment of diagnosis. The primary location most often is the adrenal gland with a small tumor size. It must be emphasized that children under 1 year of age who have metastases to the bones or distant lymph nodes should be considered as having stage 4 of the disease and should be treated more intensively, although the prognosis for such patients is much better than for older children with stage 4 of the process.
Despite the fundamentally good prognosis for babies with stage 4S, some patients may die due to respiratory disorders caused by compression of the chest by the huge liver. If there is a risk of developing respiratory disorders, low-dose liver irradiation (4-8 Gy) or low-dose chemotherapy (vincristine and cyclophosphamide) is used. In some patients (approximately 10% of cases), the process can progress to the classic stage 4 process, but most often this can be predicted by analyzing the presence of negative prognostic factors - tumor markers.
Mass screening
Determination of the level of catecholamine derivatives in the 24-hour urine of children aged 6 months is used in a number of countries for the preclinical detection of neuroblastoma. Data from Japanese researchers are the most optimistic (detection of the early stages of the disease), however, data from researchers from France, Germany, England, and the USA are more restrained, since there is an opinion that the screening method also identifies those tumors that can spontaneously regress and therefore there is a danger of “over-treatment” of small sick.
Page 192
Risk factors for neuroblastoma
A risk factor is something that increases the likelihood of developing a tumor.
Lifestyle factors are major factors in adult cancer. Examples include an unhealthy diet (low intake of fruits and vegetables), physical inactivity, smoking and drinking alcohol. Risk factors associated with children's lifestyle do not affect the occurrence of malignant tumors.
The likelihood of neuroblastoma occurring around the world is almost the same. This suggests that environmental factors, such as pollution, are not the cause of the development of this tumor.
The only known risk factor for neuroblastoma is heredity. It is believed that some people may very rarely inherit the risk of developing neuroblastoma. Only in 1-2% of cases is the familial form of neuroblastoma diagnosed, i.e. the occurrence of a tumor in a child in whose family there were cases of this disease.
The average age of patients at the time of diagnosis of familial cases of neuroblastoma is 9 months. This is less than the age of patients with sporadic (non-inherited) cases of tumor. In addition, children with familial neuroblastoma may develop two or more similar tumors in different organs, such as both adrenal glands.
It is important to distinguish neuroblastomas, which develop simultaneously in different organs, from metastatic neuroblastoma, when the tumor arises in one organ and then spreads throughout the body. If neuroblastoma occurs in several places, then a familial form of the disease can be assumed. Neuroblastoma metastases can occur in both familial and sporadic cases of the tumor.
Classification
Depending on the type and location of neuroblastoma in children, there are:
- medulloblastoma;
- retinoblastoma;
- neurofibrosarcoma;
- sympathoblastoma.
Very often this disease is confused with a malignant tumor called esthesioneuroblastoma. It is extremely rare and is classified as a separate cancer disease.
Medulloblastoma
A similar neuroblastoma in children occurs in the cerebellar region of the brain. It is not subject to surgical treatment. This form develops very quickly. Due to the enlargement of the tumor, the child’s coordination of movements is impaired.
Sympathoblastoma
Such neuroblastoma in children develops in the adrenal glands and in the tissues of the sympathetic nervous system. It occurs both in infants after birth and in the fetus in the womb of a woman in the third trimester.
Retinoblastoma
Such neuroblastoma in children is localized on the tissues of the retina. Symptoms of neuroblastoma are a sharp and causeless deterioration in vision. If the tumor continues to grow, metastases will appear in the brain.
Neurofibrosarcoma
A similar neuroblastoma in children is formed in the sympathetic department. Because of it, many metastases appear, which spread to the bones and lymph nodes.
What are the causes of neuroblastoma?
The exact causes of neuroblastoma are not clear. However, there are known differences between neuroblastoma cells and normal neuroblasts. The differences between neuroblastomas that respond to treatment and those that do not respond to therapy and have a poor prognosis (outcome) of the disease have been clarified. This information is important when developing treatment approaches.
Many researchers believe that neuroblastoma occurs when normal embryonic neuroblasts do not mature into nerve cells or adrenal cortical cells. Instead, they continue to grow and divide.
Neuroblasts may not be fully mature by the time the baby is born. In fact, it has been shown that small clusters of neuroblasts are often detected in infants up to 3 months of age. Most of these cells eventually mature into nerve cells and do not form neuroblastoma. Sometimes the neuroblasts remaining in infants continue to grow and form a tumor, which can even metastasize to various organs. However, many such tumors eventually mature or disappear.
As the child grows, the likelihood of these cells maturing decreases, and the likelihood of developing neuroblastoma increases. Once neuroblastoma reaches a large size and symptoms appear, the cells stop maturing and continue to grow and spread unless treated.
Some cancer patients have DNA mutations (changes) that they inherited from one of their parents, which increases their risk of developing a tumor. Some believe that some familial cases of neuroblastoma result from inherited mutations in a tumor-inhibiting gene.
Most neuroblastomas are not due to inherited DNA mutations. They are caused by mutations acquired early in a child's life. These mutations are present in the tumor cells of the child's parent and are not passed on to the children. The causes of DNA changes that lead to neuroblastomas are not known.
Causes
The factors that contribute to the appearance of a malignant tumor and its growth have not been established. The main causes, according to doctors and scientists, include mutations. Hereditary predisposition plays a certain role. Infectious diseases to which a woman is exposed during pregnancy also have an impact. Due to various pathological phenomena, genetic abnormalities can occur. The risk of cancer in a newborn or unborn child increases significantly if the child is exposed to carcinogens.
Is early diagnosis of neuroblastoma possible?
Studies have shown that screening (examination in the absence of symptoms of the disease) in children for the purpose of early diagnosis of neuroblastoma is of no value.
Screening (testing urine to detect certain substances) at 6 months of age has actually been able to diagnose more cases of neuroblastoma.
However, these tumors were those that either disappeared later on their own or matured and, perhaps, would never have been diagnosed.
For the same reason, it is believed that such screening will not reduce mortality from neuroblastoma.
Moreover, current screening methods are not as specific as we would like. Of the two children with suspected neuroblastoma, according to screening data, only one case actually had a tumor. These false results led to undue concern among parents and operations in children in the absence of neuroblastoma.
In rare cases, neuroblastoma can be diagnosed before birth using ultrasound (ultrasound). This method is used to determine the age of the fetus, predict the time of birth of the child and identify certain birth defects. Improvements in ultrasound and other techniques may lead to more accurate diagnosis of neuroblastoma before the baby is born.
Causes of neuroblastoma
The exact reasons that lead to the development of neuroblastoma have not been established. It is known that a tumor develops as a result of a disruption in the process of cell proliferation (division). Neuroblasts should normally transform into adult cells of the nervous system, so during their “maturation” they actively divide. When exposed to unknown causes, at some point during the division process an error occurs, and neuroblasts begin to turn into atypical cells, from which neurobalastoma is then formed.
If we talk about hereditary predisposition, which is present in many other types of cancer, then the contribution of this factor to the development of neuroblastoma is small. Only about 1% of cases of the disease are hereditary. In such patients, mutations in genes are detected, which can also lead to the development of lymphoma. The role of such common risk factors as smoking, alcohol consumption, poor environmental conditions and others in the development of neuroblastoma has not been confirmed.
How is neuroblastoma diagnosed?
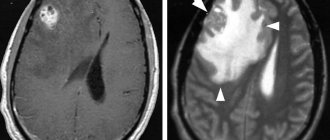
If a child has signs and symptoms suggestive of neuroblastoma, additional testing will be required, including blood and urine tests, microscopic examination of tissue, and the use of imaging techniques such as computed tomography (CT) , magnetic resonance imaging (MRI) , scanning , and etc.
This additional testing is necessary because many of the symptoms of neuroblastoma can be caused by other tumors and non-tumor diseases.
Signs and symptoms of neuroblastoma depend on the location of the primary tumor and the extent to which it has spread to nearby or distant organs and parts of the body.
The most common sign of neuroblastoma is the discovery of a tumor in the abdomen, which leads to an increase in its size. The child may complain of a feeling of abdominal distension, discomfort or pain as a result of the presence of a tumor. However, palpation of the tumor does not cause pain. The tumor may also be located in other areas, such as the neck, spreading beyond the eyeball and causing it to bulge.
Neuroblastoma often affects the bones. In this case, the child may complain of bone pain, limp, and refuse to walk. If the tumor spreads into the spinal canal, compression of the spinal cord may occur, leading to weakness, numbness and paralysis of the lower extremities.
Every fourth patient may have a fever.
Less commonly observed:
- Constant loose stools (diarrhea)
- High blood pressure leading to irritability
- Cardiopalmus
- Skin redness
- Sweating
These signs and symptoms result from the release of hormones by neuroblastoma cells.
Sometimes patients experience swelling of the lower extremities and scrotum due to compression of the blood and lymphatic vessels in the pelvic area. In some cases, a growing tumor can lead to dysfunction of the bladder and colon. Neuroblastoma's pressure on the superior vena cava, which carries blood from the head and neck to the heart, can cause swelling of the face or throat. These phenomena, in turn, can lead to breathing or swallowing problems.
As a result of pressure on nerves in the chest and neck, symptoms such as drooping eyelids and constricted pupils can occur. Compression of nerves near the spine can cause a child to lose the ability to move their arms or legs.
The appearance of bluish or reddish spots that resemble small bruises may indicate skin damage by a tumor process.
Due to the involvement of the bone marrow, which produces blood cells, a child's blood counts may all decrease, which can cause weakness, frequent infections, and increased bleeding from minor injuries (cuts or scrapes).
Normal nerve cells communicate with each other by releasing certain chemicals, the main ones being catecholamines. In the body, catecholamines are broken down into metabolites, which are then excreted in the urine.
In 90% of cases, neuroblastoma cells produce such an amount of catecholamines that can be detected in the urine using special methods. Two main metabolites of catecholamines are usually determined: homovanillic acid (HVA) and vanillylmandelic acid (VMA).
Some symptoms associated with neuroblastoma, such as high blood pressure, rapid heartbeat, and loose stools, are directly attributed to increased levels of catecholamines.
A biochemical blood test allows you to judge the function of the liver and kidneys.
Computed tomography (CT), magnetic resonance imaging (MRI), bone scans, and ultrasound (US) are used to identify the tumor and its metastases.
Although a number of examination methods suggest the presence of neuroblastoma, the final diagnosis is made only when neuroblastoma cells are detected during microscopic examination.
Sometimes neuroblastoma cells can be confused with cells from other childhood tumors. In this case, immunohistochemical examination of the tumor is used after special treatment with antibodies. These antibodies bind to neuroblastoma cells, unlike cells from other childhood tumors.
If the levels of catecholamines or their metabolites are elevated, then detection of neuroblastoma cells in the bone marrow is sufficient to establish a definitive diagnosis. The bone marrow may be affected in up to 25% of patients with neuroblastoma.
Some clinics are using a new scanning method using radioactive metaiodobenzylguanidine (MYBG). The method allows you to detect neuroblastoma cells in the bone marrow and other parts of the body.
Stages
At the first stage of neuroblastoma in children, the tumor is no more than 5 mm. Metastases do not appear during this period. This neuroblastoma rarely shows symptoms, making the disease very difficult to diagnose.
At the second stage of nephroblastoma in children, the tumor increases to 1 cm. The adjacent lymph nodes are gradually affected.
At the third stage of neuroblastoma, symptoms will already be clearly visible. The tumor is actively growing and affecting organs on the other side of the body. The tumor can grow up to 10 cm.
At the fourth stage, signs of neuroblastoma in children also appear due to the fact that cancer cells move to more distant areas of the body. At this stage, several tumors may appear in different parts of the body at once.
Treatment of neuroblastoma in children
Every child with neuroblastoma should receive treatment. The treatment method depends on the stage of the tumor, the age of the child, and prognostic factors. Treatment includes surgery, chemotherapy and/or radiation therapy. In some children, two or all three therapies may be used.
Usually, when choosing a treatment method for neuroblastoma, they take into account not so much the stage of the disease as the risk group.
Low risk group. In general, low-risk children require only surgery, with the exception of some patients. For this small group of children, chemotherapy may be used.
Intermediate risk group. Patients with this risk group are prescribed 4-8 cycles of chemotherapy before or after surgery. In some cases, repeated surgery is performed to remove the remaining tumor or radiation therapy.
High risk group. For this group of patients, very intensive chemotherapy is used along with bone marrow or peripheral stem cell transplantation.
Surgery and/or radiation therapy may be part of the overall treatment program. Biological agents, such as 13-cis-retinoid acid, are prescribed for 6 months after discontinuation of therapy.
Unfortunately, some patients after treatment for neuroblastoma may experience a recurrence (recurrence) of the disease after initial treatment. Treatment in this case depends on many factors, including risk group and location of relapse, and may include surgery and chemotherapy.
Diagnostic methods
In order to make an accurate diagnosis, assess possible risks and plan treatment, the patient must undergo a comprehensive examination. It may include the following methods:
- Lab tests. Neuroblastoma markers include neuron-specific enolase (NSE) and catecholamine metabolites. To obtain general information about the patient’s health, routine tests are prescribed - a general blood test, biochemical study, etc.
- In order to exclude or confirm the presence of metastases in the bone marrow, a puncture is performed followed by immunohistochemical and morphological assessment.
- Biopsy and histological examination are key in making a diagnosis. The stage of neuroblastoma is determined using the Hughes and INPC systems.
- To visualize the primary tumor and distant metastases, ultrasound, CT, MRI, PET-CT, and scintigraphy are prescribed.
- Molecular genetic studies are aimed at determining the expression of key neuroblastoma genes.
There are two ways to make an accurate diagnosis. The first is the exact histological picture, which speaks in favor of neuroblastoma. The second is an increase in the level of tumor markers in combination with the presence of tumor cells in the bone marrow.