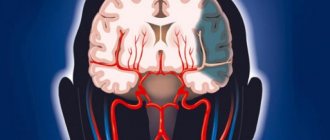
Hallervorden-Spatz disease (progressive rigidity) is a rare hereditary neurodegenerative disease that affects the basal ganglia and accumulates iron in them.
Cumulative processes occur in the substantia nigra and globus pallidus. Also in the cerebral cortex, changes in nerve cells are observed, the appearance of pigmented neuroglia, and glial cells similar to glia in Alzheimer's disease.
The disease was first described by German researchers Julius Hallervorden and Hugo Spatz, after whom it was named.
However, in modern international medical practice, the disease is called neurodegeneration with brain iron deposition (NBIA). The abandonment of the original name is due to the fact that Julius Hallervorden conducted research on euthanasia in Nazi Germany.
The disease is considered rare, with up to 3 people having the disease per 1 million people. This neurodegeneration can lead to parkinsonism, dystonia, dementia, and death.
Features of genetic failure
Hallervorden-Spatz disease is a hereditary genetic pathology.
The disease can be either familial or sporadic (random), but it is always inherited. The disease belongs to the autosomal recessive type of inheritance. To transmit the disease, both parents must be heterozygous carriers of the disease and have one mutated allele.
The disease is caused by a mutation in the pantothenate kinase gene (PANK2), which is located on chromosome 20 at the 20p12.3-p13 locus. The pantothenate kinase gene is responsible for encoding the protein pantothenate kinase 2, which in turn inhibits the accumulation of the amino acids cysteine and panthetheine.
As a result, chemical compounds with iron ions are formed, which have a negative effect on proteins and trigger peroxidation processes, which leads to programmed cell death of neurons. Instead of dead neurons, glial tissue (auxiliary cells of nervous tissue) grows.
Pathological processes occur in the globus pallidus, red nuclei and substantia nigra (substantia nigra). A greenish-brown substance that contains iron accumulates in them. In addition, spheroid neuroaxonal formations (extensions of axons with proliferation of tubular and membranous structures) are observed in the white matter of the brain, cerebral cortex, peripheral nerve trunks and spinal cord.
2. Reasons
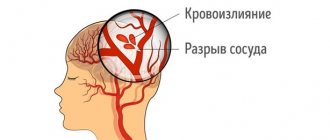
To date, questions remain regarding pathogenesis. According to the most reasoned and generally accepted point of view, the characteristic clinical picture arises due to the abnormal, irregular structure of the tissues from which the walls of microvessels are formed at the stage of intrauterine development: they have practically no elastic muscle fibers, and the shell of the endothelial wall is connective tissue. With this composition, the vascular wall is fragile, vulnerable and unstable to stretching; it easily forms aneurysms and ruptures, which leads to bleeding.
Visit our Cardiovascular Surgery page
Features of the clinical picture
Signs of this pathology may vary in each specific case, and the manifestation of symptoms depends on the form of the disease in the individual.
The childhood form of Hallervorden Spatz appears between the ages of 5 and 10 years. It is considered a classic form of the disease.
In the vast majority of cases (90%), the disease begins with torsion dystonia, which affects the leg muscles. The patient experiences muscle contraction, difficulty walking, and changes in gait. Then the muscles of the face, pharynx and trunk are affected.
The presence of blepharospasm, hand spasm, spasmodic torticollis, and facial hemispasm may be present. A third of patients have muscle rigidity, hypokinesia, and tremor (signs of Parkinsonism syndrome).
Typical signs of the childhood form of the disease also include:
- epileptic syndrome;
- aggressiveness;
- mental retardation (occurs due to deterioration of memory and attention);
- asociality;
- visual disturbances (optic atrophy, retinal degeneration);
- mental disorders.
The childhood form of the disease progresses quickly, and complete loss of the ability to move occurs 10-15 years after the onset of the disease.
The teenage form develops and progresses more slowly. At the beginning of the disease the following appear:
- focal torsion dystonia (most often affects the muscles of the limbs and maxillo-oral muscles);
- neuropsychological disorders;
- behavioral disorders;
- intellectual disorders.
The atypical form of the disease is the rarest (15% of cases), and proceeds differently from the first two forms. The most typical symptoms for this form are:
- Parkinsonism syndrome with the inability to maintain balance while standing;
- involuntary movements in different parts of the body, dystonia, chorea and athetosis (slow and jerky movements), hemiballismus (sweeping movements), myoclonus (non-prolonged muscle spasms);
- dementia;
- epileptic syndrome;
- depression, aggression;
- pathological reflexes that arise as a result of destructive processes in the neurons of the brain.
Eponyms in medicine are the designations of diseases, syndromes, symptoms and reflexes named after their discoverer(s). Eponymous names are quite widely used in medicine - with their help it is easier to describe a particular clinical phenomenon. They certainly help specialists understand each other better. In the pedagogical process, their use makes it easier to master the material, facilitating memorization.
In some countries, eponymous names are used very widely in medical practice, while in others their use is quite limited. The history of eponymous names in medicine is not simple; there have been cases when the original name underwent changes or completely disappeared from the medical lexicon.
In principle, whether or not to use eponyms is a matter of taste, with one exception - if they do not raise ethical issues [1]. Thus, when the use of a particular eponymous name for a disease or syndrome is associated with a violation of ethical standards by the discoverer, its use seems immoral, therefore the eponym should be deleted from practical activities and medical literature [1]. This primarily applies to persons whose scientific activities were of a particularly cruel nature, or who themselves participated in crimes against humanity. Moreover, it does not matter whether the eponymous name was given BEFORE the commission of the corresponding crimes [1].
Unfortunately, eponymous names in honor of people who participated in Nazi crimes remain in medical practice to this day. In connection with this, the literature naturally raises the question of the need to remove them from the doctor’s vocabulary [1-6].
The above, however, does not mean a complete rejection of eponyms. They must be used, especially for the purpose of perpetuating the memory of people who actively fought against Nazism or who died innocently, whose only fault was that they simply lived in that difficult time.
Before World War II, German medicine was rightfully considered one of the most developed in the world [2]. By 1939, there were only 9 Nobel laureates in Germany [3]. How did it happen that it was specialists in the field of neuroscience who participated, often with enthusiasm, in the inhumane projects of the Nazis? There are many reasons for this: some wanted quick career growth, some wanted to gain recognition from the scientific community, some wanted to receive additional subsidies, but the main one was permissiveness, which allowed them to engage in “pure science” “for the good of the state.” In this regard, it is very symbolic that among German doctors there was the highest percentage (45%) of members of the Nazi Party and SA (storm troops of the Nazi Party) - 26% [4], and among SS members there were 7 times more doctors than in the general population - 7% and less than 1%, respectively [4, 5]. Moreover, often after the Second World War, these people remained unpunished [6]. Thus, out of at least 350 German doctors who participated in inhumane experiments, the Nuremberg Tribunal in 1946-1947. brought to justice only 23 [3].
In medicine, some eponymous names are still actively used, the origin of which is associated with German doctors who were former Nazis or who actively collaborated with them. Let's look at some of them.
Hallerwarden-Spatz disease
In 1921, J. Hallervorden, together with H. Spatz, showed that excessive iron deposition in the area of the globus pallidus and the reticular part of the substantia nigra causes the development of muscle rigidity (pantothenate kinase-related neurodegeneration).
The description of this disease was based on observations of 5 sisters from one family with 12 children; 3 children died in childhood, and 4 were clinically healthy. The remaining 5 sisters, aged 7-9 years, developed a disease that manifested itself as gait disturbances with muscle rigidity in the legs, foot deformities and dystonia. The progression of the disease was manifested by intellectual decline and dysarthria; in 2 cases muscle atrophy of the lower extremities developed and in 1 case corticospinal symptoms developed, in 1 case choreoathetosis and swallowing disorders developed; hyperpigmentation of the skin was characteristic. Death in all cases of the disease occurred at the age of 16-27 years.
J. Hallervorden, who observed the patients during their lifetime, went to Munich, taking with him the brain of one of the deceased girls (cited from [7]). There he met H. Spatz, together with whom a description of the disease was made and under the eponymous name “Hallervorden-Spatz disease” this disease remains to this day [1, 7], although in rare cases the name “Martha-Alma disease” is used - by the names of the sisters whose brains were examined [7].
Let us now dwell in more detail on the specialists who described the disease.
Julius Hallervorden (1882-1965) from January 1, 1938 worked as a professor and head of the department of neuromorphology at the Institute for Brain Research. Kaiser Wilhelm in Berlin1.
He was one of the doctors who actively collaborated with the Nazis [2, 8]. He himself admitted that the brain samples he studied during the Nazi period in power in Germany were taken from victims of euthanasia [1, 2, 7]. There is evidence [1] that he participated in the murder of more than 60 children and adolescents in Brandenburg on October 28, 1940, and even reported that he personally isolated the brains of euthanasia victims. There is also evidence [2, 8] that Hallervorden personally examined living people, choosing among them those who were about to die. The phrase he said after the war, full of cynicism, is known: “The main thing is that I received brain drugs. Where they come from and how they get to me is none of my business” [1]. Hallervorden's collection consisted of 697 brain samples obtained by himself and 2097 that he collected from various centers where the euthanasia program was carried out [3]. In 12 works published by J. Hallervorden after the Second World War, data obtained from these collections were used [2, 7]. In 1948, he continued to work as head of the neuromorphology department at the Institute. Max Planck and, moreover, became the chairman of the German Society of Neuromorphologists and remained so until his resignation [2, 7].
It should be noted that, under public pressure in the post-war period, a number of German universities, including the prestigious Institute for Brain Research. Max Planck, removed all the remains of Nazi victims from his collections (including the “collection” of euthanasia victims of J. Hallervorden), cremating them [4].
Hugo Spatz (1888-1969) is a famous German psychiatrist. In 1937, he was appointed director of the Institute for Brain Research. Kaiser Wilhelm in Berlin, replacing O. Vogt, who was dismissed from this position by the Nazis [1]. Together with Hallervorden, Spatz was involved in carrying out a "highly productive scientific study" based on the killings of children as part of the euthanasia project [1, 2]. Under the leadership of Spatz, this institute worked closely with the institute in Brandenburg-Gorden, from where it received hundreds of brain preparations from mentally ill people [1]. Towards the end of the war, he moved most of his “pathological collection” to Munich [1]. After the war, H. Spatz was arrested by the American military police precisely as the director of the Institute for Brain Research. Kaiser Wilhelm. However, he was never imprisoned and was even invited to work at the American Aeromedical Center in Heidelberg, where he continued his research [1]. Later, despite the crimes, he was offered a job in the physiological laboratory at the Institute in Hesse, where he conducted research on the hypothalamus and pineal gland. Later he moved to the Institute. Max Planck, where he continued his research on the primate brain [1].
Taking into account the above, it is proposed not to use the term “Hallervorden-Spatz disease” in the scientific and practical activities of doctors and instead use the terms “Martha-Alma disease”, “neurodegeneration with accumulation of iron in the brain” or “neuroaxonal dystrophy”.
Van Bogart-Scherer-Epstein syndrome
In 1937, Ludo van Bogaert, Hans Joachim Scherer, and Emil Epstein described 2 patients with generalized cholesterolosis of the lungs, tendons, and central nervous system. The disease manifested itself as diffuse damage predominantly to the central nervous system in the form of mental retardation, behavioral disorders, dementia, pyramidal symptoms (up to severe para- or tetraparesis), cerebellar ataxia, bulbar disorders, epileptic seizures in combination with tendon xanthoma (especially the Achilles tendon) and juvenile cataracts [9-11]. Thirty years later, a defect in the synthesis of bile acids leading to cholesterol accumulation was identified, and subsequently a genetic defect in this rare autosomal recessive disease was identified [12].
Of the three authors who described the disease, G.I. was associated with the Nazis. Scherer.
Hans Joachim Scherer (1906–1945) was a German physician and neuromorphologist known for his work in the field of morphology and biology of malignant gliomas [1, 3]. However, few people know about the participation of G.I. Scherer in the Nazi euthanasia project [1, 2]. During World War II he worked at the Neurological Institute in Breslau (Silesia). It was there that G.I. Scherer was directly involved in the pathomorphological analysis of approximately 300 brain specimens from Polish and German children subjected to the euthanasia program at the child psychiatric clinic near Loben [2, 3]. Moreover, he did this not under duress, but voluntarily, being one of the active participants in this inhumane program [1].
It is proposed in scientific and practical activities instead of the term “Van Bogaert-Scherer-Epstein syndrome” to use the already widely used term “cerebrotendinous xanthomatosis” [1-3].
Scholz-Bielschowsky-Henneberg disease
Willibald Oscar Scholz (1889-1971) in 1925 described 3 children with progressive leukodystrophy, whose clinical and pathomorphological data were similar to metachromatic leukodystrophy (due to methodological difficulties, metachromasia was not identified then). Considering Scholz’s close collaboration with Bielschowsky and Henneberg, one of the forms of metachromatic dystrophy received the eponymous name “Scholz-Bilschowsky-Henneberg disease” [3].
IN. Scholz defended his dissertation in Jena in 1914 and subsequently worked as a chief specialist at a psychiatric and neurological clinic in Leipzig, then at the German Research Institute of Psychiatry in Munich, and after the death of W. Spielmeyer became the director of this institute and remained so until 1961 This institute was actively involved in the T4 Program and has studied at least 194 brain samples from euthanasia victims and published at least 11 papers based on the findings.
It is proposed not to use the term “Scholz-Bilschowsky-Henneberg disease”, but instead to use the term “Bilschowsky-Henneberg disease”.
Other doctors who actively collaborated with the Nazis
In total, about 400,000 people have been subjected to forced sterilization in Germany since 1933 [3]. Among them, 80–96% were people with “congenital dementia,” schizophrenia and congenital epilepsy [3]. Also included in this program were patients with bipolar disorder, Huntington's disease, large brain malformations, congenital blindness, congenital severe hearing impairment, chronic alcoholism, presenile and senile dementia, encephalitis, polio, “treatment-resistant paralysis,” multiple sclerosis, disease Parkinson [3]. Pediatricians, psychiatrists and neurologists were required to report the presence of such patients to the relevant authorities, and all anatomical departments in Nazi Germany, without exception, used the bodies of people tortured by the Nazis [13].
After the outbreak of World War II in September 1939, the Nazis concluded that an active euthanasia program was of greater economic benefit than sterilization, since the former freed up hospital beds for wounded soldiers [3]. During 1939-1941. The Nazis carried out both a child euthanasia program and an adult euthanasia program (Action T4 program) [3]. As a result, more than 70 thousand adults and children were killed, mainly by gas, starvation or lethal injections [3]. Despite the fact that this program was officially terminated after protests by relatives of the victims and the intervention of the Catholic Church, unofficially it continued until 1945, claiming the lives of 275 thousand people [3].
Thus, the German neurologist Georg Schaldenbrand (G. Schaltenbrand, 1897-1979), working at the university hospital in Würzburg, conducted a series of experiments on people - without the consent of them or their relatives - to study the possible viral pathogenesis of multiple sclerosis [3]. He administered intraventricular cerebrospinal fluid from patients with definite multiple sclerosis to monkeys, and then from monkeys to 45 mentally ill people (it is known [3] that in 2 cases this led to death).
The Austrian pediatric neurologist Heinrich Gromm (H. Gross, 1915–2005) performed euthanasia at the Spiegelgrund Hospital in Vienna, where he collected a large collection of brain samples from children undergoing euthanasia [2, 3]. He subsequently published a series of articles on this material, including co-authorship with former SS member, neurologist and pathologist Franz Seitelberger [3]. The latter continued to publish with H. Gross even in the 80s, when it became known that the latter was a war criminal [2]. At the same time, F. Seitelberger in the 70s was the rector of the University of Vienna, an honored member of various international societies, and even received prestigious scientific awards [2].
What happened in the 30s and 40s of the last century is not a purely German peculiarity. Few people know that there were active followers of eugenics and euthanasia in other countries. Thus, a number of fairly well-known doctors of various specialties from different countries, who were not members of the Nazi Party, supported eugenics and euthanasia and also left their mark on medicine in the form of eponymous names. Among them, it should be noted the French pediatrician E.Ch. Apert (E. Apert), Swiss internist and cardiologist W. His (W. His), Anglo-American neurologist, Irish by birth, R.F. Kennedy (R. Kennedy) and the American geneticist M.T. M. Macklin [1, 14]. The question of whether or not to preserve eponymous names in these cases is also very actively debated [2].
The materials presented in this article give reason to agree with the proposals expressed in the literature to change eponyms associated with Nazism to other, simpler and more understandable names of diseases and syndromes [1]. Of course, the list can be continued, and here doctors of different specialties could provide invaluable assistance.
M. Bielschowsky, who was dismissed under the “Law on the Restoration of the Professional Civil Service” (in other words, the law on racial purity), worked in this post until 1933.
Diagnostic criteria and techniques
Making a diagnosis of Hallervorden-Spatz disease can be problematic for neurologists due to the polymorphism of symptoms.
Main criteria for diagnosis:
- onset of illness before age 30;
- typical picture in MRI results;
- constant development of symptoms;
- extrapyramidal syndromes;
- pyramid signs;
- epileptic seizures;
- cognitive impairment;
- retinal pigmentary degeneration;
- presence of the disease in a family history.
“Eyes of the tiger” on MRI with contrast is a typical picture of Hallervorden Spatz disease.
The disease must be differentiated from other diseases that have a similar clinical picture: Huntington’s chorea, Wilson-Konovalov disease, choreoacanthocytosis, Machado-Joseph disease.
For diagnosis, the patient is referred for the following examinations:
- Study of neurological status by a neurologist, neuropathologist.
- Electroencephalography.
- Consultation with an ophthalmologist . Direct ophthalmoscopy and visual acuity testing are possible.
- Genetic studies to determine the type of inheritance.
- DNA studies (detection of mutations in the PANK2 gene).
- Positron emission tomography of the brain . It is carried out to detect decreased blood circulation in the frontoparietal lobes, striatum, and globus pallidus.
- Magnetic resonance imaging of the brain . Required to detect the "eye of the tiger" - a hyperintense oval area in the hypointense zone that occurs due to iron accumulation. There is no single theory about the development of the “eye of the tiger”; there are assumptions about its occurrence before the onset of clinical manifestations of the disease, and about its appearance years after the onset of the disease. MRI can also detect spheroid neuroaxonal formations.
Pathogenesis
In most patients, especially with early manifestations of the disease, mutations are detected in the gene encoded by the enzyme pantoten kinase (PANK2), on chromosome 20p13. This enzyme plays a crucial role in the biosynthesis of coenzyme-A. The enzyme defect leads to the accumulation of cysteine, which in the presence of iron (which means especially in the substantia nigra and basal ganglia) leads to an increase in free radicals and contributes to oxidative damage to the brain. The overall process leads to the deposition of iron and neuromelanin.
How can modern doctors help?
There are no treatments in modern medicine that can prevent or stop Hallervorden-Spatz disease.
Therapy is aimed at alleviating and relieving the intensity of symptoms:
- For parkinsonism syndrome, dopamine agonists (Pronoran, Pramipexole, Mirapex, Piribedil) and amantadines (Simmetrel, Midantan) are prescribed. However, the syndrome is often resistant to treatment.
- To relieve hyperkinesis, valproates (Konvulex, Depakin, Encorat) and benzodiazepines (Clonazepam, Diazepam) are used.
- To relieve muscle spasticity, muscle relaxants (Mydocalm, Baclofen) are used.
- Epileptic seizures are relieved with valproate , Tomapax.
- For cognitive impairment, Gliatilin and Neuromidin .
- For the treatment of mental disorders, it is recommended to take antipsychotics (Clonazepam, Quetiapine, Rispolent), antidepressants (Dapoxetine, Citalopram, Venlafaxine).
New methods of treating the disease are also emerging. These include therapy by administering pantothenic acid, magnetic stimulation of the brain (globus pallidus).
Sad, but not hopeless
With Hallervorden-Spatz disease, symptoms constantly progress.
The childhood form of the disease is the most severe. Complete disability of an individual occurs 10-15 years from the moment of the first appearance of clinical manifestations. The most favorable development of the disease is predicted in the adult form of the disease. Especially in cases of mild dementia.
Therapy allows the patient to maintain the relative quality of life and his ability to self-care. Life expectancy with the atypical form of Hallervorden-Spatz disease can be more than 20 years.
4.Treatment
The primary goals are to prevent dangerous hemorrhages and relieve anemia; in some cases, blood transfusions are prescribed for this purpose. For a long time, nasal tamponade was considered the therapeutic standard for Randu-Osler disease, but now more and more specialists are abandoning this method, finding a lot of shortcomings in it and low efficiency. Today, among the therapeutic agents used are irrigation with a solution of aminocaproic acid, various coagulation techniques (cryo-, electro-, laser) and artificial embolization of the most problematic arteries. The presence of large aneurysms often forces them to resort to surgical removal.
Patients with hereditary hemorrhagic telangiectasia have to follow a special diet and take all precautions to avoid injury to bleeding and potentially dangerous lesions.