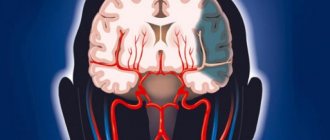
Болезнь Галлервордена-Шпатца (ригидность прогрессирующая) – редкое наследственное нейродегенеративное заболевание, при котором происходит поражение базальных ганглиев, и накопление в них железа.
Накопительные процессы происходят в черной субстанции и в бледном шаре. Также в коре головного мозга наблюдаются изменения в нервных клетках, появление пигментированных нейроглий, и глиальных клеток, сходных с глиями при болезни Альцгеймера.
Впервые болезнь была описана немецкими исследователями Юлиусом Галлерворденом и Гуго Шпатцем, именами которых и была названа.
Однако в современной международной медицинской практике болезнь называют нейродегенерацией с отложением железа в головном мозге (NBIA). Отказ от первоначального названия связан с тем, что Юлиус Галлерворден вел исследования эвтаназии в фашистской Германии.
Заболевание считается редким, на 1 миллион человек приходится до 3 человека, имеющих болезнь. Данная нейродегенерация может приводить к паркинсонизму, дистонии, деменции, смерти.
Особенности генетического сбоя
Болезнь Галлервордена-Шпатца это наследственная генетическая патология. Заболевание может быть как семейным, так и спорадическим (случайным), однако всегда передается по наследству. Болезнь относится к аутносомно-рецессивному типу наследования.
Для передачи болезни оба родителя должны быть гетерозиготными носителями заболевания и иметь одну мутировавшую аллель.
Болезнь вызывается мутацией в гене пантотенат-киназы (PANK2) который находится в 20 хромосоме в локусе 20р12.3–р13. Ген пантотенат-киназы отвечает за кодирование протеина пантотенат-киназы 2, который в свою очередь сдерживает процесс накопления аминокислоты цистеина и пантетеина.
В результате происходит образование химических соединений с ионами железа, которые имеют негативное воздействие на белки и дают старт процессам перекисного окисления, что приводит к программируемой клеточной гибели нейронов. Вместо отмерших нейронов нарастает глиальная ткань (вспомогательные клетки нервной ткани).
Патологические процессы происходят в бледном шаре, красных ядрах и черном веществе (субстанции nigra). В них скапливается субстанция зеленовато-коричневого цвета, которая содержит железо. Помимо этого в белом веществе головного мозга, коре головного мозга, периферических нервных стволах и спинном мозге наблюдаются сфероидные нейроаксональные образования (расширения аксонов с разрастанием тубулярных и мембранных структур).
2.Причины
На сегодняшний день в отношении патогенеза остаются вопросы. Согласно наиболее аргументированной и общепринятой точке зрения, характерная клиническая картина возникает вследствие аномального, неправильного строения тканей, из которых на этапе внутриутробного развития формируются стенки микрососудов: в них практически нет эластичных мышечных волокон, а оболочкой эндотелиальной стенки служит соединительная ткань. При таком составе сосудистая стенка отличается хрупкостью, уязвимостью и неустойчивостью к растяжениям, она легко образует аневризмы и разрывы, – что и приводит к кровотечениям.
Посетите нашу страницу Сердечно-сосудистая хирургия
Особенности клинической картины
Признаки данной патологии могут варьироваться в каждом конкретном случае, и проявление симптомов зависит от формы болезни у индивида.
Детская форма Галлервордена Шпатца проявляется в возрасте между 5 и 10 годами. Считается классической формой болезни.
В подавляющем количестве случаев (90%) заболевание начинается с торсионной дистонии, которая поражает мышцы ног. У больного происходит сокращение мышц, затрудняется ходьба, изменяется походка. Затем поражаются мышцы лица, глотки и туловища.
Возможно присутствие блефароспазма, спазма кистей, спастической кривошеи, лицевого гемиспазма. Треть пациентов имеют мышечную ригидность, гипокинезию, тремор (признаки синдрома паркинсонизма).
К типичным признакам детской формы болезни также относят:
- эпилептический синдром;
- агрессивность;
- умственную отсталость (возникает из-за ухудшения памяти и внимания);
- асоциальность;
- зрительные нарушения (атрофия зрительных нервов, дегенерация сетчатки);
- психические расстройства.
Детская форма заболевания прогрессирует быстро, и полная потеря способности к передвижению наступает через 10-15 лет после дебюта болезни.
Подростковая форма развивается и протекает более медленно. В начале болезни проявляются:
- фокальная торсионная дистония (чаще всего затрагивает мышцы конечностей и челюстно-ротовые мышцы);
- нейропсихологические расстройства;
- поведенческие расстройства;
- интеллектуальные расстройства.
Атипичная форма болезни – наиболее редкая (15% случаев заболевания), и протекает отлично от первых двух форм. Для этой формы наиболее характерны такие симптомы:
- синдром паркинсонизма с невозможностью сохранять равновесие в положении стоя;
- непроизвольные движения в разных частях тела, дистония, хорея и атетоз (замедленные и порывистые движения), гемибаллизм (размашистые движения), миоклония (не длительные мышечные спазмы);
- деменция;
- эпилептический синдром;
- депрессивность, агрессия;
- патологические рефлексы, которые возникают вследствие разрушительных процессов в нейронах головного мозга.
Эпонимы в медицине — это обозначение заболеваний, синдромов, симптомов и рефлексов, названных в честь их первооткрывателя (ей). Эпонимические названия довольно широко используются в медицине — с их помощью легче дать описание того или иного клинического явления. Они, безусловно, лучше помогают специалистам понять друг друга. В педагогическом процессе их применение делает более легким усвоение материала, облегчая запоминание.
В некоторых странах эпонимические названия в медицинской практике используются очень широко, а в других — их применение довольно ограничено. История эпонимических названий в медицине не проста, бывали случаи, когда первоначальное название претерпевало изменения или совсем исчезало из медицинского лексикона.
В принципе, использовать или нет эпонимы — дело вкуса, за одним исключением — если они не затрагивают вопросы этики [1]. Так, когда использование того или иного эпонимического названия заболевания или синдрома связано с нарушением этических норм первооткрывателем, его применение представляется аморальным, поэтому эпоним следует вычеркнуть из практической деятельности и медицинской литературы [1]. Это в первую очередь относится к лицам, чья научная деятельность носила особенно жестокий характер, либо они сами участвовали в преступлениях против человечности. Причем не имеет значения, было ли эпонимическое название дано ДО совершения соответствующих преступлений [1].
К сожалению, до наших дней в медицинской практике остаются эпонимические названия в честь людей, участвовавших в преступлениях нацистов. В литературе в связи с этим закономерно ставится вопрос о необходимости их удаления из лексикона врача [1—6].
Сказанное выше тем не менее не означает полного отказа от эпонимов. Их необходимо использовать, особенно с целью увековечивания памяти активно боровшихся с нацизмом или безвинно погибших людей, вина которых заключалась лишь в том, что они просто жили в то непростое время.
До Второй мировой войны немецкая медицина по праву считалась одной из самых развитых в мире [2]. Одних только Нобелевских лауреатов к 1939 г. в Германии насчитывалось 9 человек [3]. Как же произошло, что именно специалисты в области нейронаук участвовали, нередко с энтузиазмом, в бесчеловечных нацистских проектах? Причин для этого много: кто-то хотел быстрого карьерного роста, кто-то — добиться признания научного сообщества, кто-то — получить дополнительное субсидирование, но основная из них — вседозволенность, позволявшая «во благо государства» заниматься «чистой наукой». В связи с этим весьма символично выглядит тот факт, что среди немецких врачей был самый высокий процент (45%) членов нацисткой партии и СА (штурмовые отряды нацисткой партии) — 26% [4], а среди членов СС врачей было в 7 раз больше, чем в общем населении — соответственно 7% и менее 1% [4, 5]. Причем нередко после Второй мировой войны эти люди оставались безнаказанными [6]. Так, из по меньшей мере 350 участвовавших в бесчеловечных экспериментах немецких врачей Нюрнбергский трибунал в 1946—1947 гг. привлек к ответственности лишь 23 [3].
В медицине до сих пор активно используются некоторые эпонимические названия, происхождение которых связано с немецкими врачами, бывшими нацистами или активно с ними сотрудничавшими. Остановимся на некоторых из них.
Болезнь Галлервордена—Шпатца
В 1921 г. Ю. Галлерворден совместно с Х. Шпатцем показали, что избыточное отложение железа в области бледного шара и ретикулярной части черного вещества вызывает развитие мышечной ригидности (пантотенаткиназасвязанная нейродегенерация).
В основу описания этой болезни были положены наблюдения 5 сестер из одной семьи, в которой было 12 детей; 3 ребенка умерли в детском возрасте, а 4 — были клинически здоровы. У остальных 5 сестер в возрасте 7—9 лет манифестировало заболевание, проявившееся нарушениями ходьбы с мышечной ригидностью в ногах, деформацией стоп и дистонией. Прогрессирование болезни проявлялось интеллектуальным снижением и дизартрией; в 2 случаях развилась атрофия мышц нижних конечностей и в 1 — кортикоспинальная симптоматика, в 1 случае развились хореоатетоз и нарушения глотания; характерной была гиперпигментация кожных покровов. Летальный исход во всех случаях заболевания наступил в возрасте 16—27 лет.
Наблюдавший при жизни пациенток Ю. Галлерворден уехал в Мюнхен, взяв с собой головной мозг одной из умерших девочек (цит. по [7]). Там он и встретил Х. Шпатца, совместно с которым было сделано описание болезни и под эпонимическим названием «болезнь Галлервордена—Шпатца» это заболевание остается до сих пор [1, 7], хотя в редких случаях используется название «болезнь Марты—Альмы» — по именам сестер, головной мозг которых был исследован [7].
Остановимся теперь более подробно на описавших болезнь специалистах.
Юлиус Галлерворден (Julius Hallervorden, 1882—1965 гг.) с 1 января 1938 г. работал профессором и руководителем отдела нейроморфологии в Институте по изучению мозга им. Кайзера Вильгельма в Берлине1.
Он являлся одним из врачей, активно сотрудничавших с нацистами [2, 8]. Сам он признавал, что образцы головного мозга, которые изучались им в период нахождения нацистов у власти в Германии, были взяты у жертв эвтаназии [1, 2, 7]. Есть данные [1], что он участвовал в убийстве более чем 60 детей и подростков в Бранденбурге 28 октября 1940 г. и даже рапортовал, что лично выделял головной мозг у жертв эвтаназии. Существуют также свидетельства [2, 8] того, что Галлерворден лично осматривал живых людей, выбирая среди них тех, кто должен был умереть. Известна сказанная им после войны полная цинизма фраза: «Главное, я получал препараты головного мозга. А откуда они и как они попадают ко мне — это не мое дело» [1]. Коллекция Галлервордена состояла из 697 образцов головного мозга, полученных им самим, и 2097 — которые он собрал в различных центрах, где проводилась программа эвтаназии [3]. В 12 работах, опубликованных Ю. Галлерворденом уже после Второй мировой войны, применялись данные, полученные при использовании этих коллекций [2, 7]. В 1948 г. он продолжил работу в должности руководителя отделения нейроморфологии в Институте им. Макса Планка и, более того, стал председателем Германского общества нейроморфологов и был им вплоть до отставки [2, 7].
Следует заметить, что под давлением общественности в послевоенное время ряд немецких университетов, включая престижный Институт по исследованию головного мозга им. Макса Планка, удалил все останки жертв нацистов из своих коллекций (включая и «коллекцию» жертв эвтаназии Ю. Галлервордена), кремировав их [4].
Хьюго Шпатц (Hugo Spatz, 1888—1969 гг.) — известный немецкий психиатр. В 1937 г. он был назначен директором Института по изучению мозга им. Кайзера Вильгельма в Берлине, сменив уволенного с этой должности нацистами О. Фогта [1]. Вместе с Галлерворденом Шпатц участвовал в выполнении «высокопродуктивного научного исследования», основанного на убийствах детей в рамках проекта эвтаназии [1, 2]. Под руководством Шпатца указанный институт работал в тесном контакте с институтом в Бранденбурге—Гордене, откуда получал сотни препаратов головного мозга психически больных людей [1]. Ближе к концу войны он перевез большую часть своей «патоморфологической коллекции» в Мюнхен [1]. После войны Х. Шпатц был арестован американской военной полицией именно как директор Института по изучению мозга им. Кайзера Вильгельма. Однако он никогда не находился в заключении и даже был приглашен на работу в американский Аэромедицинский центр в Гейдельберге, где продолжал исследования [1]. Позднее, несмотря на преступления, ему была предложена работа в физиологической лаборатории в Институте в Гессене, где им были проведены исследования гипоталамуса и шишковидной железы. В дальнейшем он перешел в Институт им. Макса Планка, где продолжил исследования головного мозга приматов [1].
Учитывая изложенное выше, предлагается термин «болезнь Галлервордена—Шпатца» в научной и практической деятельности врачей не использовать и вместо него применять термины «болезнь Марты—Альмы», «нейродегенерация с накоплением в головном мозге железа» либо «нейроаксональная дистрофия».
Синдром Ван Богарта—Шерера—Эпштейна
В 1937 г. Людо ван Богарт (L. Van Bogaert), Ганс Иоахим Шерер (H. Scherer) и Эмиль Эпштейн (E. Epstein) описали 2 пациентов с генерализованным холестеринозом легких, сухожилий и ЦНС. Заболевание проявлялось диффузным поражением преимущественно ЦНС в виде задержки психического развития, поведенческих расстройств, деменции, пирамидной симптоматики (вплоть до выраженного пара- или тетрапареза), мозжечковой атаксии, бульбарных нарушений, эпилептических приступов в сочетании с ксантомой сухожилий (особенно ахиллово сухожилие) и ювенильной катарактой [9—11]. Спустя 30 лет был выявлен приводящий к накоплению холестерина дефект синтеза желчных кислот, а в дальнейшем — и генетический дефект этого редкого аутосомно-рецессивного заболевания [12].
Из трех описавших болезнь авторов с нацистами был связан Г.И. Шерер.
Ганс Иоахим Шерер (Hans Joachim Scherer, 1906—1945 гг.) — немецкий врач и нейроморфолог, известный работами в области морфологии и биологии злокачественных глиом [1, 3]. Однако мало кто знает об участии Г.И. Шерера в нацистском проекте эвтаназии [1, 2]. Во время Второй мировой войны он работал в Неврологическом институте в Бреслау (Силезия). Именно там Г.И. Шерер был непосредственно вовлечен в патоморфологический анализ около 300 препаратов головного мозга польских и немецких детей, подвергшихся программе эвтаназии в детской психиатрической клинике около Лобена [2, 3]. Причем делал он это не по принуждению, а добровольно, будучи одним из активных участников этой бесчеловечной программы [1].
Предлагается в научной и практической деятельности вместо термина «синдром Ван Богарта—Шерера—Эпштейна» использовать уже достаточно широко применяющийся термин «церебротендинозный ксантоматоз» [1—3].
Болезнь Шольца—Бильшовского—Хеннеберга
Виллибальд Оскар Шольц (Willibald Oscar Scholz, 1889—1971 гг.) в 1925 г. описал 3 детей с прогрессирующей лейкодистрофией, клинические и патоморфологические данные которых были сходны с метахроматической лейкодистрофией (в силу методических трудностей метахромазия тогда не была выявлена). Учитывая тесное сотрудничество Шольца с Бильшовским и Хеннебергом, одна из форм метахроматической дистрофии получила эпонимическое название «болезнь Шольца—Бильшовского—Хеннеберга» [3].
В.О. Шольц защитил диссертацию в Йене в 1914 г. и в дальнейшем работал главным специалистом в психиатрической и неврологической клинике в Лейпциге, затем — в Немецком исследовательском институте психиатрии в Мюнхене, а после смерти W. Spielmeyer стал директором этого института и был им вплоть до 1961 г. Этот институт активно участвовал в осуществлении «Программы Т4», и в нем было изучено не менее 194 образцов головного мозга жертв эвтаназии, а на основании полученных данных было опубликовано не менее 11 статей.
Предлагается термин «болезнь Шольца—Бильшовского—Хеннеберга» не использовать, а вместо него применять термин «болезнь Бильшовского—Хеннеберга».
Другие активно сотрудничавшие с нацистами врачи
Всего в Германии с 1933 г. было подвергнуто насильственной стерилизации около 400 000 человек [3]. Среди них 80—96% составляли лица с «врожденным слабоумием», шизофренией и врожденной эпилепсией [3]. Также в эту программу были включены пациенты с биполярным расстройством, болезнью Гентингтона, крупными мальформациями головного мозга, врожденной слепотой, врожденными выраженными нарушениями слуха, хроническим алкоголизмом, пресенильной и сенильной деменцией, энцефалитами, полиомиелитом, «резистентными к лечению параличами», рассеянным склерозом, болезнью Паркинсона [3]. Педиатры, психиатры и неврологи должны были в обязательном порядке сообщать о наличии таких больных в соответствующие органы, а все без исключения анатомические отделения в нацистской Германии использовали тела замученных нацистами людей [13].
После начала Второй мировой войны в сентябре 1939 г. нацистами был сделан вывод о «большей экономической выгоде» активной программы эвтаназии по сравнению со стерилизацией, поскольку первая позволяла освобождать места в госпиталях для раненых солдат [3]. В течение 1939—1941 гг. нацисты проводили как детскую программу эвтаназии, так и взрослую (программа «Action T4») [3]. В результате было умерщвлено более 70 тыс. взрослых и детей, преимущественно газом, голоданием или летальными инъекциями [3]. Несмотря на то что официально эта программа была прекращена после протестов родственников погибших и вмешательства католической церкви, неофициально она продолжалась вплоть до 1945 г., унеся жизни 275 тыс. человек [3].
Так, немецкий невролог Георг Шальденбранд (G. Schaltenbrand, 1897—1979 гг.), работая в университетском госпитале в Вюрцбурге, провел серию экспериментов на людях — без согласия их или их родственников — по изучению возможного вирусного патогенеза рассеянного склероза [3]. Он проводил внутрижелудочковое введение цереброспинальной жидкости от пациентов с достоверным рассеянным склерозом обезьянам, а затем от обезьян 45 психически больным людям (известно [3], что в 2 случаях это привело к летальному исходу).
Австрийский детский невролог Хейнрих Громм (H. Gross, 1915—2005 гг.) проводил эвтаназию в госпитале Spiegelgrund в Вене, где собрал большую коллекцию образцов головного мозга детей, подвергшихся эвтаназии [2, 3]. В дальнейшем он опубликовал серию статей на этом материале, в том числе в соавторстве с бывшим членом СС, неврологом и патоморфологом Францем Сейтельбергером (F. Seitelberger)[3]. Последний продолжал публиковаться с H. Gross даже в 80-е годы, когда стало известно о том, что последний является военным преступником [2]. При этом Ф. Сейтельбергер в 70-е годы был ректором Венского университета, заслуженным членом различных международных обществ и даже получал престижные научные награды [2].
То, что случилось в 30—40-е годы прошлого века, не является чисто немецкой особенностью. Мало кто знает, что активные последователи евгеники и эвтаназии были и в других странах. Так, ряд довольно известных врачей различных специальностей из разных стран, не являясь членами нацистской партии, поддерживали евгенику и эвтаназию и также оставили свой след в медицине в виде эпонимических названий. Среди них следует отметить французского педиатра Е.Ч. Аперта (E. Apert), швейцарского интерниста и кардиолога В. Хиса (W. His), англо-американского невролога, ирландца по происхождению, Р.Ф. Кеннеди (R. Kennedy) и американского генетика М.Т. Маклина (M. Macklin) [1, 14]. Вопрос о том, сохранять или нет эпонимические названия в этих случаях также весьма активно дискутируется [2].
Приведенные в настоящей статье материалы дают основание согласиться с высказываемыми в литературе предложениями изменить связанные с нацизмом эпонимы на другие, более простые и понятные названия заболеваний и синдромов [1]. Разумеется, их список можно продолжить, и здесь неоценимую помощь могли бы оказать врачи разных специальностей.
На этом посту до 1933 г. работал M. Bielschowsky, уволенный по «Закону о восстановлении профессиональной гражданской службы» (иными словами, закон о расовой чистоте).
Диагностические критерии и методики
Постановка диагноза «болезнь Галлервордена-Шпатца» может быть проблематичной для неврологов из-за полиморфизма симптомов.
Основные критерии для диагностики:
- начало болезни до 30 лет;
- типичная картина в результатах МРТ;
- постоянное развитие симптомов;
- экстрапирамидальные синдромы;
- пирамидные знаки;
- эпилептические приступы;
- когнитивные нарушения;
- пигментная дегенерация сетчатки;
- наличие болезни в семейном анамнезе.
«Глаза тигра» на МРТ с контрастом — типичная картина при болезни Галлервордена Шпатца
Болезнь необходимо дифференцировать с другими заболеваниями, которые имеют сходную клиническую картину: хореей Гентингтона, болезнью Вильсона-Коновалова, хореоакантоцитозом, болезнью Мачадо-Джозефа.
Для диагностирования пациент направляют на такие обследования:
- Изучение неврологического статуса у невролога, невропатолога.
- Электроэнцефалография.
- Консультация офтальмолога. Возможно проведение прямой офтальмоскопии, проверки остроты зрения.
- Генетические исследования, для определения типа наследования.
- Исследования ДНК (обнаружение мутаций в гене PANK2).
- Позитронная эмиссионная томография головного мозга. Проводится для обнаружения снижения кровообращения в лобно-теменных долях, стриатуме, бледном шаре.
- Магнитно-резонансная томография головного мозга. Требуется для обнаружения «глаза тигра» — гиперинтенсивного овального участка в гипоинтенсивной зоне, который возникает из-за скопления железа. Не существует единой теории о развитии «глаза тигра», есть предположения о его возникновении до начала клинических проявлений болезни, и о его появлении через годы после начала течения заболевания. Также МРТ позволяет обнаружить сфероидные нейроаксональные образования.
Патогенез
У большинства пациентов, особенно при ранней манифестации заболевания, определяются мутации в кодируемом фермент пантотенкиназу (PANK2) гене, в хромосоме 20p13. Этот фермент играет решающую роль в биосинтезе кофермента-А. Дефект фермента ведет к накоплению цистеина, который в присутствии железа (что означает — в особенности в области чёрного вещества и базальных ганглиев) ведет к повышению свободных радикалов и способствует оксидатному повреждению мозга. Процесс в целом приводит к отложению железа и нейромеланина.
Чем могут помочь современные врачи?
В современной медицине не существует методов лечения, которые могут предотвратить или остановить болезнь Галлервордена-Шпатца.
Терапия направлена на облегчение и снятие интенсивности симптоматики:
- При синдроме паркинсонизма назначают агонисты дофамина (Проноран, Прамипексол, Мирапекс, Пирибедил), амантадины (Симметрел, Мидантан). Однако часто наблюдается резистентность синдрома к лечению.
- Для купирования гиперкинезов используют вальпроаты (Конвулекс, Депакин, Энкорат), бензодиазепины (Клоназепам, Диазепам).
- Для снятия спастичности мышц применяются миорелаксанты (Мидокалм, Баклофен).
- Эпилептические приступы снимаются вальпроатами, Томапаксом.
- При когнитивных нарушениях применяют Глиатилин, Нейромидин.
- Для лечения психических нарушений рекомендуют прием нейролептиков (Клоназепама, Кветиапина, Рисполента), антидепрессантов (Дапоксетина, Циталопрама, Венлафаксина).
Появляются и новейшие методы лечения болезни. К ним относятся терапия путем введения пантотеновой кислоты, магнитная стимуляция головного мозга (бледного шара).
Печально, но не безнадежно
При болезни Галлервордена-Шпатца симптомы постоянно прогрессируют. Наиболее тяжело протекает детская форма заболевания. Полная инвалидизация индивида наступает через 10-15 лет с момента первого проявления клинических проявлений.
Наиболее благоприятное развитие болезни прогнозируется при взрослой форме болезни. Особенно в случае слабой выраженности деменции.
Терапия позволяет сохранить относительное качество жизни пациента и его способность к самообслуживанию. Продолжительность жизни при атипичной форме болезни Галлервордена-Шпатца может составлять более 20 лет.
4.Лечение
Первоочередными задачами являются предотвращение опасных геморрагий и купирование анемии, – в ряде случаев с этой целью назначают переливания крови. Длительное время терапевтическим стандартом при болезни Рандю-Ослера считалась тампонада носа, однако в настоящее время все больше специалистов отказываются от этого метода, находя в нем массу недостатков при малой эффективности. Сегодня в числе лечебных средств применяется орошение раствором аминокапроновой кислоты, различные методики коагуляции (крио-, электро-, лазерная) и искусственной эмболизации наиболее проблемных артерий. Наличие крупных аневризм зачастую вынуждает прибегать к хирургическому их устранению.
Пациентам с наследственной геморрагической телеангиэктазией приходится соблюдать особую диету и соблюдать все меры предосторожности, чтобы избегать травмирования кровоточивых и потенциально опасных очагов.