Medical information is reliable Checked by Shaidullin Renat Flyurovich
Creutzfeldt-Jakob disease is a rare type of degenerative brain disease. It is part of a group of prion transmissible encephalopathies, which are caused by the entry of an abnormal isoform of the prion protein into the body and its accumulation in neurons.
The pathology occurs in one in a million people on the globe. The disease was first described in 1920 by the German neurologist Hans Creutzfeldt. And a year later, his fellow countryman and colleague Alfons Jacobs clarified that the deviation is characterized by the manifestation of mental disorders against the background of organic damage to the pyramidal, extrapyramidal region of the central nervous system, the anterior part of the horns of the spinal cord, the cerebellum and visual impairment associated with changes in cortical structures.
The incidence of this pathology is low - it is registered in one person per million. In the 90s of the last century, symptoms of a new variant of Creutzfeldt-Jakob disease began to be noted, which were of an infectious nature and were associated with infection from cows and other cattle (“mad cow disease”). Most often, the disease is registered in people over the age of 65, but in recent years, deviations of this type have also been observed in younger patients.
What causes Creutzfeldt-Jakob syndrome
The disease is caused by the presence in the body of a special protein (prion), which has a disrupted structure and has a negative effect on the nervous system. The action of a prion is reminiscent of the effect of viruses that cause cell death during their reproduction, but it does not carry genetic material (DNA or RNA).
In its desire to get rid of the aggressor, the cell that is being attacked begins to actively release substances. But the protein deposited on the membrane does not allow them to come out, and they destroy the organelles and trigger the process of apoptosis. Neighboring structures begin to react to this with inflammation, and therefore further cell damage occurs.
Prions can withstand a wide temperature range, radiation, and are resistant to enzymatic degradation. They are not affected by antibiotics and antivirals. Under the influence of this protein, the tissue of the nervous system becomes “leaky” and begins to resemble a sponge.
Exogenous infection (in addition to spontaneous internal transformation of prions and the hereditary form of the disease) occurs from the external environment in the following ways:
- When eating food of animal origin. Especially a lot of the infectious agent “mad cow disease” is observed in beef, brain, blood and spleen. Cases of the disease are described among peoples who practice ritual cannibalism.
- When transplanting internal organs, including liver, kidneys, cornea, heart muscle. The possibility of transmission remains during transfusion of both whole blood and its individual components (mass of red blood cells, leukocytes, plasma).
- The use of drugs obtained from animal tissue.
- Transmission from person to person through biological fluids by getting them onto wound surfaces or into the stomach.
Creutzfeldt-Jakob disease is not transmitted by airborne droplets, as is the case with viral infections, or by ordinary touch. There are suggestions of transmission of the pathological prion protein through sputum, urine and feces, but there has been insufficient research into this variant of infection.
Prognosis of Creutzfeldt-Jakob disease
In almost all cases, this disease leads to the death of the patient within one year. Sometimes death occurs earlier - six months after the diagnosis was made, but in especially rare cases - in approximately 5% of cases - the duration of the disease can increase to two years. 10% of sick people do not survive even a few weeks, which is caused by the individual characteristics of their bodies.
Since the disease is not transmitted by airborne droplets, wearing masks and other protective equipment is not mandatory for medical workers and relatives in contact with patients. Those employees who conduct analyzes of biological fluids and tissues of patients suspected of having Creutzfeldt-Jakob disease must follow safety rules and work exclusively with gloves. In particular, direct contact of mucous membranes with samples of contaminated material should be avoided. If such extremely undesirable contact does occur, it is imperative to disinfect with a 4% hydroxide solution. Treatment is carried out within 5-10 minutes after contact, followed by rinsing the mucous membrane with running water.
Disinfection of instruments is carried out by autoclaving for an hour at a temperature of 132°C or by sterilizing for an hour in a 10% sodium hypochlorite solution or 4% sodium hydroxide solution. Standard methods of sterilizing instruments, such as formaldehyde, are ineffective in this case.
Pathogenesis of the disease
Currently, there are two types of pathogenesis of Creutzfeldt-Jakob disease. The first is extracerebral, which does not relate to brain structures and cerebral. Both types occur when infected with prions from the outside, and the hereditary and sporadic forms of the disease are still insufficiently studied.
The extracerebral variant of the development of the pathology is that the distorted protein enters the body and combines with lymphocytes, macrophages and other cells involved in immune defense. It is transported by them to the tissues of the lymphatic system (spleen, lymph nodes). There it multiplies, and the body does not interfere with this, since it does not perceive the prion as a foreign body. At the same time, it does not damage or cause enlargement of lymphoid organs, but only multiply and persist in them.
Subsequent spread of the pathogen occurs under the influence of the autonomic nervous system. They are sent to the central nervous system, and the cerebral stage of Creutzfeldt-Jakob disease begins. They accumulate in intracellular fluid and nerve endings. The result is a decrease in normal prion protein, which is involved in normal cell nutrition.
1.General information
The situation with prion proteins, the discovery of which by S. Prusiner’s group (1982) was subsequently awarded the Nobel Prize, is in many ways reminiscent of the revolution in scientific and, in particular, medical thinking, which was produced somewhat earlier by the discovery of viruses. As in the case of viruses, it was possible to explain the mechanism of development of a number of diseases, until then known only at a hypothetical level. As with viruses, fundamental scientific discussions are still ongoing: whether to consider prions as a special, transitional form of life or as a rare biochemical phenomenon.
Prions are a special type of protein that is capable of precisely reduplicating its molecules by transforming other proteins with a similar structure. Thus, when prions enter living tissue, its structure gradually changes: biochemical reactions continuously occurring at the molecular level increase the content of the prion fraction and reduce the number of intact, undamaged cells. As a result, parenchyma specialized to perform certain functions is usually the holy of holies of the central nervous system, i.e. the substance of the brain, as well as other neural tissues, turns into something like a spongy-porous dead organic mass, which gives the name to this entire group of neurodegenerative processes: transmissible spongiform (spongy) encephalopathy.
Prion diseases affect both animals (just remember the large-scale outbreaks of epidemics of mad cow disease, sheep scabies, etc.) and humans. Creutzfeldt-Jakob disease is not the only one, but the most famous and widespread of human spongiform prion encephalopathies (over 80%), which was first described about 100 years ago and received a comprehensive explanation only with the discovery of prion proteins.
Like other known forms of neurodegeneration, Creutzfeldt-Jakob disease is currently incurable, but its peculiarity is a much more rapid and malignant course: 80-90% of patients die within the first year from the onset of the disease, and only a few survive more than two years from the onset of the disease. manifestations. The problem is complicated by the fact that prion proteins are extremely stable and, unlike pathogenic microorganisms, retain their destructive properties both during conventional sterilization and under intense physical influences (for example, hard radiation).
A must read! Help with treatment and hospitalization!
Classification of Creutzfeldt-Jakob disease
Depending on the characteristics of the symptoms, the following forms are distinguished in Creutzfeldt-Jakob disease:
- subacute encephalopathy with diffuse tissue damage and rapid progression;
- dyskinetic, or classic (a combination of extrapyramidal and pyramidal disorders with manifestations of dementia);
- intermediate with a predominance of symptoms of damage to the cerebellum and subcortical areas;
- amyotrophic (with speech and movement disorders).
According to etiology, Creutzfeldt-Jakob disease is divided into the following types:
- Spontaneous. The second name is classic. In this case, prions do not enter the body from the outside, but are formed for no apparent reason. Most likely, modern medical science simply cannot find an explanation for this phenomenon. Older people (after 50 years) are more often affected.
- Hereditary. With such a deviation, the formation of a prion is genetically determined.
- Iatrogenic. In this type of disease, the damaging protein enters the body from the outside. Previously, some drugs could serve as its source. Most of them are currently not in use. The cause is also the membranes taken from the tissue of corpses to close wounds during brain surgery.
- A new form of Creutzfeldt-Jakob disease. First described by UK doctors in 1995. The reason for this was the consumption of meat from “mad cows” containing prions. Unlike the classic version, the pathology is observed in people over the age of 20 and manifests itself in the form of personality changes.
4.Treatment
Therapy for Creutzfeldt-Jakob disease today, always and everywhere (including widely advertised Western clinics) is symptomatic, purely palliative. As far as possible, the phenomena of motor and coordination insufficiency are eliminated or mitigated, drugs are prescribed that can to some extent inhibit the decay of cognitive functions (for example, nootropics), reduce the frequency of epileptic attacks, and correct behavioral and affective disorders.
However, a strategy for a truly etiopathogenetic treatment that would block the reproduction of prion molecules by pharmacological or the body's own immune agents has yet to be developed. In research centers of developed countries, significant efforts are being made in this direction, and they are bringing the first results from those that are usually cautiously called encouraging in science.
Creutzfeldt-Jakob disease: symptoms
Creutzfeldt-Jakob disease has characteristic symptoms, regardless of origin. Iatrogenic, hereditary and classical forms manifest themselves in approximately the same way. The new type of disease proceeds somewhat differently.
Main manifestations
The initial stage is manifested by temporary loss of memory of events, changes in mood, and loss of interest in others. Gradually, the patient experiences more and more difficulties in carrying out vital actions. The final stage is accompanied by blurred vision, slow speech and the appearance of hallucinations.
In approximately 40% of patients with the sporadic form, the disease progresses subacutely, with gradually worsening cognitive impairment. Another 40% of those infected suffer from dysfunction of the cerebellum, and the rest have a mixed form of the disease.
The most characteristic clinical symptoms of classical Creutzfeldt-Jakob disease are:
- conduct disorder;
- memory impairment;
- diplopia and development of cortical blindness;
- mental disorders;
- cerebellar dysfunction, ataxia;
- lack of coordination;
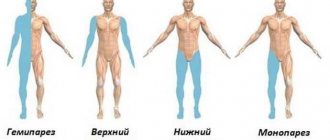
- paresis;
- dysarthria;
- inability to self-care, sloppiness and other signs of dementia;
- a combination of signs of damage to the pyramidal and extrapyramidal systems.
Many patients experience local or generalized types of epileptic seizures. They appear in response to certain stimuli - light, sound, touch.
The terminal stage is accompanied by global cognitive impairment and complete inability to move. Death usually occurs within 8 months to 2 years after the onset of the initial clear symptoms of the disease. The new form of the disease is characterized by mental disorders and changes in sensitivity that come to the fore.
Stages
Experts distinguish three stages of Creutzfeldt-Jakob disease:
- Prodromal stage. Its symptoms do not have specific manifestations and are observed in approximately 30% of all patients. The person experiences asthenia, sleep and appetite disturbances, decreased memory ability, weight loss, and mild cognitive impairment. The patient is characterized by the appearance of phobias, delusions, and hallucinations.
- Initial stage. Changes in vision begin, headache appears, instability, ataxia, sensory disturbances, tremor, myoclonus, epileptic seizures, and other neurological symptoms increase against the background of dementia.
- The advanced stage is accompanied by spastic paralysis, all signs of disturbances in the functioning of brain structures, and a severe form of dementia without the possibility of contact with the patient. A person completely loses control over the functioning of the pelvic organs, swallowing is impaired, and a decrease in temperature is noted.
Most often, Creutzfeldt-Jakob disease develops gradually, but sometimes it has an acute and subacute onset. Death occurs as a result of failure of vital centers and severe exhaustion.
3. Symptoms, diagnosis
Classic, iatrogenic and hereditary types are characterized by a rapid increase in behavioral and cognitive (memory, attention, etc.) disorders, polymorphic epileptic seizures, disorders of coordination of movements, vision, muscle tone. The first stages of the “new variant” of Creutzfeldt-Jakob disease are characterized by the predominance of affective (emotional-volitional) disorders - depressive, apathetic-abulic - as well as loss of body weight and the phenomenon of ataxia (impaired coordination of movements and spatial orientation).
However, it is important to understand that all forms are dementing (weakening), accompanied by rapidly and steadily progressing neurological and psychiatric symptoms, loss of the ability for basic self-care, and result in death.
Clear clinical and diagnostic criteria have been developed and applied for Creutzfeldt-Jakob disease in all its forms. Additional examination usually includes electroencephalography, tomographic imaging methods, neuropsychological diagnostics, specific blood and cerebrospinal fluid tests; in some cases and only with informed consent, a biopsy of the brain substance is performed. However, the most complete and unambiguous answers are provided only by pathomorphological post-mortem examination.
About our clinic Chistye Prudy metro station Medintercom page!
Diagnosis of Creutzfeldt-Jakob disease
A presumptive diagnosis of Creutzfeldt-Jakob disease is made based on clinical manifestations and additional research methods. Pathology can be suspected if there are a combination of certain symptoms observed over a long period of time:
- progression of dementia;
- pyramidal and extrapyramidal disorders;
- myoclonus;
- disorders of the cerebellar structures;
- visual impairment.
To clarify the pathology, the neurologist refers the person to undergo encephalography, MRI of the brain, spinal puncture, stereotactic biopsy:
- On the EEG, almost every patient with Creutzfeldt-Jakob disease shows a decrease in electrical activity with periodic sharp waves. Changes characteristic of myoclonus are observed in 50% of patients. A new type of disease often does not manifest itself as any abnormalities on the encephalogram.
- MRI can reveal signs of atrophic changes in the cerebellum and cerebral cortex (honeycomb syndrome), and dilatation of the ventricles. Areas with increased signal are sometimes detected from subcortical structures and the thalamus.
- Lumbar puncture is mandatory if Creutzfeldt-Jakob disease is suspected. The study of cerebrospinal fluid allows for a differential diagnosis with other pathological disorders of the central nervous system.
- The most objective way to reliably determine the diagnosis is a morphological examination of a biopsy of brain tissue. Using the immunochromatographic method, it is possible to detect the prion protein. Its presence suggests that the patient has Creutzfeldt-Jakob disease.
Differential diagnosis must be made with other diseases of the central nervous system that occur with similar symptoms:
- senile, multi-infarct dementia;
- Alzheimer's disease;
- Hashimoto's encephalopathy;
- inflammatory damage to the meninges (encephalitis, arachnoiditis, meningitis).
Treatment of Creutzfeldt–Jakob disease
In modern medicine there are no ways to successfully cure patients from this disease. In recent decades, scientists have tested many drugs: antibiotics, steroids, immunomodulators and antiviral drugs. But all the methods considered showed a complete lack of therapeutic effect.
Today, when working with patients suffering from Creutzfeldt-Jakob disease, doctors can provide exclusively symptomatic treatment aimed at alleviating the severity of the disease, but not at destroying prions and improving the brain. If CBJ is suspected, the patient is taken off medications that negatively affect the mnestic functions of his body.
A positive effect on the condition of patients, namely relief of symptoms of the disease, was identified with the use of the following medications: Brefeldin A and calcium channel blockers. The former, by destroying the Golgi apparatus, create an obstacle to the production of PrPSc in the structure of infected cells. The latter stop the functioning of NMDA receptors, which causes a longer lifespan of infected neuronal cultures.
Treatment of Creutzfeldt-Jakob disease in Moscow
Etiotropic therapy for the disease is still under development. Therefore, all treatment in the clinic is aimed at relieving symptomatic manifestations. All previously used medications are discontinued for the patient, since they can negatively affect the course of the disease, exacerbating the disturbance of mnestic functions and worsening the patient’s behavior.
Conservative therapy
It has already been proven that traditional means that are successfully used in the treatment of viral infections (vaccines, interferons) do not have a positive effect on the course of Creutzfeldt-Jakob disease. Some results with a temporary improvement in condition can be obtained by introducing an antibiotic, which blocks the work of the Golgi apparatus inside the cell and prevents the proliferation of prions. It has been noted that when using certain calcium channel blockers, the duration of functioning of the affected brain structures increases over time.
A slight slowdown in the accumulation of pathological proteins is observed when using an immunomodulator, which stimulates the production of endogenous interferon. But these drugs have strong side effects.
Symptomatic therapy in patients is aimed at eliminating:
- tremors and epileptic seizures;
- extrapyramidal disorders;
- depression and anxiety;
- pain syndrome.
For this purpose, various groups of drugs are used, their choice is made by the doctor. The basis for the use of a particular remedy is the presence and severity of the deviation. The following groups of drugs are used in therapy:
- neuroleptics;
- tranquilizers;
- analgesics;
- opiates;
- sedatives;
- antidepressants;
- anticonvulsant medications.
The advantage of having the patient in the clinic
After a diagnosis of Creutzfeldt-Jakob disease is made, a person needs to provide full symptomatic therapy, psychological support for the patient and his family members. The further the pathology progresses, the more care and attention the patient needs, as he gradually loses self-care skills and the functioning of the pelvic organs is disrupted.
Working relatives cannot provide round-the-clock vigil near the patient. And he will need to be fed, washed and helped with movement as needed. In some cases, he has to remove urine through catheterization as a result of bladder dysfunction. And this manipulation can only be performed by specialists with medical education.
If swallowing function is impaired, a person will need nutrition using a special tube and drip solutions with the necessary substances. At home, it is possible to provide full treatment and care only in the earliest stages. Subsequently, the patient will definitely require systematic care from specially trained personnel.
In the terminal stage, measures are taken to prolong life. They are carried out only in the presence of resuscitation equipment - mechanical ventilation, oxygen therapy, means for stimulating the respiratory and cardiovascular systems.
When choosing a clinic to provide care to patients with Creutzfeldt-Jakob disease, relatives should understand that the highest quality care can be provided in an institution that employs experienced specialists with a specific profile.
If you hospitalize a person in a public hospital, you can save money, but the conditions of stay will not be ideal:
- several people are accommodated in the wards;
- you will come to purchase medications on your own, since those that are available cannot always effectively eliminate complications;
- the patient is at high risk of contracting a hospital-acquired infection;
- staff will not be able to constantly monitor the patient and provide assistance if necessary 24 hours a day.
When the patient is still conscious and able to assess the current situation, his stay in a government institution will significantly aggravate the condition, cause depression and increased mental disorders.
Questions and answers
If you are in constant contact with a person who has Creutzfeldt-Jakob disease, is there a chance of becoming infected?
Infection through normal contact is extremely rare. But during care you should adhere to hygiene rules. They include washing your hands before eating, protecting your skin from body fluids, and covering all wound surfaces with waterproof bandages.
Is it possible to cure a person with this disease?
This disease cannot be treated and is fatal with a 100% probability. All that can be done in this case is to provide decent care for the patient, stop emerging complications and carry out symptomatic therapy in order to improve the quality of life.
Why does Creutzfeldt-Jakob disease cause death?
As the pathology progresses, various complications develop, which most often become the cause of death. Some of them are amenable to drug correction - epileptic seizures, acute psychoses, pneumonia due to infection. Proper care can reduce a person's suffering from pressure sores resulting from muscle degeneration. The final outcome of the disease is the development of respiratory or cardiovascular failure, which ends in death. But even these functions can be maintained for some time with the help of special devices.