Резкое двустороннее снижение остроты зрения может говорить о развитии опасной патологии — оптической нейропатии Лебера. Такое заболевание чаще затрагивает молодых людей мужского пола. Рассмотрим причины патологии, симптомы и особенности терапии в статье.
Впервые случаи заболевания, при котором острота зрения стремительно снижается из-за атрофии зрительного нерва, были описаны в 1871 году Теодором Лебером. Ученый наблюдал за развитием патологии у молодых людей из нескольких родственных семей, что указывало на наследственную причину заболевания. Механизм наследования болезни был тщательно изучен, поэтому удалось доказать, что патология передается по материнской линии преимущественно мужчинам.
До сих пор характер заболевания недостаточно хорошо изучен для того, чтобы удалось найти эффективные методы лечения такой болезни. Сейчас лечение больных заключается в поддерживающей терапии, но периодически группы ученых представляют новые методики генной терапии для клинических исследований, что, конечно, вселяет надежду людям с подобным диагнозом.
Оптическая нейропатия Лебера: симптомы, лечение болезни
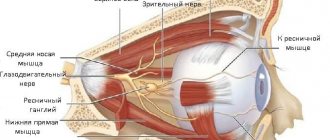
Любые нарушения зрения, стремительно прогрессирующие, должны быть тщательно изучены и комплексно продиагностированы. Оптическая нейропатия Лебера редко сопровождается недомоганием, характерным для неврологических заболеваний, при этом зрение падает быстро, иногда даже не в течение нескольких месяцев, а за две-три недели. При такой остроте, зрение невозможно проверить по таблице Сивцева, потому что человек попросту не видит знаков, поэтому применяют аппаратную диагностику и метод «счета пальцев» на определенном расстоянии от лица пациента. Особенность заболевания в том, что зрительный нерв поражается из-за мутации гена. Вырабатывается избыточное количество токсичных молекул кислорода, что негативно сказывается на состоянии клеток нерва и зрение снижается.
Оптическая нейропатия Лебера — симптомы и особенности болезни:
- молодой возраст пациентов — 18-35 лет;
- болезнь чаще развивается у мужчин;
- быстрая односторонняя, а затем и двусторонняя потеря зрения;
- острота зрения снижается в течение нескольких недель;
- нарушение цветового восприятия красного и зеленого цветов;
- патология может сопровождаться симптомами, присущими митохондриальным болезням (судороги, нарушение проводимости сердца).
При атрофии зрительного нерва Лебера чаще всего процесс развития болезни начинается с одного глаза, но нередки случаи, когда поражаются сразу два глаза. Центральное зрение снижается постоянно. Это связано с постепенной смертью клеток зрительного нерва, передающего информацию об изображении в головной мозг. Иногда болезнь может сопровождаться симптомами, характерными для неврологических заболеваний. В этом случае больной человек страдает от дистонии, тремора, атаксии. Реже в начале развития патологии человек замечает искры, пятна, яркие вспышки перед глазами.
Атрофия зрительных нервов Лебера: клинико-генетические аспекты (научный обзор)
Л.Г. Кириллова, А.А. Шевченко, Л.Ю. Силаева, ГУ «Институт педиатрии, акушерства и гинекологии АМН Украины», г. Киев
Резюме
Излагаются современные взгляды на проблему митохондриального заболевания — атрофии зрительных нервов Лебера, оценка различных методов диагностики и лечения, а также необходимость проведения молекулярно-генетического исследования для обнаружения патогенных мутаций митохондриальной ДНК. Своевременная диагностика позволяет избежать ненужных обследований для реализации клинической эффективности проводимого лечения и прогноза течения заболевания.
Ключевые слова
атрофия зрительных нервов Лебера, митохондриальное заболевание, молекулярно-генетическое исследование.
В последние годы настолько изменилась классическая клиника многих заболеваний в нейропедиатрии, что опытный клиницист часто мысленно ставит себе вопрос: «Что же все-таки представляет клиника и течение патологического процесса, с которым я сталкиваюсь?». Порой очень трудно на консилиуме врачей прийти к единому мнению по поводу того или иного заболевания, и эта трудность во многих случаях может быть объяснена тем, что мы, зная о существовании ряда митохондральных заболеваний, не имеем возможности ни подтвердить, ни исключить предполагаемую патологию без проведения углубленных генетических исследований.
На практике приходится сталкиваться с тем, когда к педиатру, а затем к неврологу обращаются родители с детьми, которые прогрессивно теряют остроту зрения с офтальмологическими изменениями на глазном дне. В таких случаях приходится прежде всего исключать разного рода воспалительные процессы, опухоли мозга и наследственно-дегенеративные заболевания. Редко мы думаем о митохондральной атрофии зрительных нервов Лебера. Поводом для написания этой статьи послужили два случая заболевания детей, которые находились в отделении детской психоневрологи ИПАГ АМН Украины в течение последнего года (у одного из них диагноз врожденной атрофии Лебера выставлен в г. Москве и подтвержден генетически).
Атрофия зрительных нервов Лебера известна как семейная форма слепоты с конца XIX века, когда в 1871 году немецкий офтальмолог Теодор Лебер дал первое описание этой патологии. Данное заболевание проявляется быстрым двусторонним снижением зрения с центральными скотомами, расстройством цветового зрения и наследуется по рецессивному, сцепленному с полом типу. Учитывая, что основным субстратом поражения является зрительный нерв, а во многих случаях имеются и другие неврологические нарушения и поражения, болезнь Лебера может быть отнесена к неврологическим заболеваниям [3, 29]. Следует отметить и тот факт, что зрительная атрофия Лебера является болезнью, при которой были обнаружены наследственные точечные мутации митохондриальной ДНК. В 1988 году D.C. Wallace et al. было установлено, что атрофия зрительных нервов Лебера связана с заменой 11778-го нуклеотида митохондриального гена, кодирующего 4-ю субъединицу комплекса I дыхательной цепи. В последующем были обнаружены и другие мутации, ведущие к этой болезни. В большинстве случаев они затрагивают митохондриальные гены, кодирующие белки, которые участвуют в переносе электронов в дыхательной цепи с нарушением фосфорилирующей функции митохондрий. В 95 % случаев в митохондриальной ДНК выявляются три мутации в 11778, 3460 и 14484-м положениях [4, 15, 17]. Мутация 11778А (замена оснований гуанин/аденил в положении 11778 митохондриальной ДНК, приводящая к замене аргинина на гистидин в 340-м кодоне гена субъединицы 4 NADH-дегидрогеназы), встречается в 69 % случаев почти во всех популяционных группах. Мутация 14484С (замена оснований тиамин/цитозин в положении 14484С митохондриальной ДНК, приводящая к замене метионина на валин в гене субъединицы 6 NADH-дегидрогеназы), встречается в 14 % случаев, концентрируясь у выходцев из Нидерландов и Англии, а мутация 3460А (замена оснований гуанин/аденин в положении 3460 митохондриальной ДНК, приводящая к замене аланина на треонин в 52-м кодоне гена субъединицы 1 NADH-дегидрогеназы) — приблизительно в 13 % случаев. Мутация 15275 была найдена в меньшинстве семей. Митохондриальные мутации, ведущие к болезни Лебера, передаются от матери всем детям, однако заболевание развивается преимущественно у сыновей. Установлено, что соотношение пораженных мужчин и женщин соответственно составляет 5 : 1, а средний возраст манифестации — от 23 до 26 лет (самое раннее начало в 4 года и самое позднее — в 86 лет), врожденные формы отмечаются преимущественно у детей. Также было установлено, что наиболее часто данная патология встречается у жителей Северной Европы или японцев, причем имеются межпопуляционные различия в соотношении первичных мутаций. Например, в Азии доля мутации 11778А выше, чем в странах Запада [13, 29, 35]. Согласно эпидемиологическим данным, частота болезни Лебера в северо-восточной Англии составляет 3,3 на 105 населения, частота носительства — 8,9 на 105, а в Финляндии ее распространенность — около 1 : 50000 [6, 14]. В Австралии среди всех зарегистрированных слепых больные с атрофией зрительных нервов Лебера составляют 0,4-2 % [19]. В числе внешних факторов, которые повышают пенетрантность мутантного гена и провоцируют манифестацию болезни Лебера, называют употребление алкоголя и курение. При этом влияние курения связывают с действием цианидов, окиси углерода и других токсинов табачного дыма на процессы окислительного фосфорилирования. Также имеются отдельные сообщения о провоцирующих внешних факторах в виде черепно-мозговых травм, психоэмоционального напряжения, андрогенных препаратов, противотуберкулезного препарата этамбутола и противовирусных препаратов, применяемых при СПИДе [10, 12, 22, 30]. Описываемые клинические случаи болезни Лебера представлены как обширными родословными с большим числом больных лиц, так и спорадическими случаями атрофии зрительных нервов при отсутствии указания на характерный семейный анамнез. Вызывает интерес проведенное канадскими генетиками из университетов Квебека и Монреаля исследование, которое позволило определить происхождение болезни Лебера в Квебеке. Им удалось выявить девушку, которая передала своим потомкам — жителям Квебека эту редкую генетическую болезнь. Ею оказалась одна из 700 девушек-сирот, которых французский король Людовик ХIV в 1663-1673 годах отправил в Прекрасную провинцию (как тогда называли Квебек) для улучшения и коренного изменения сложившейся там демографической ситуации. Используя обширную базу данных обо всех квебекцах, родившихся после 1800 года, была установлена генеалогия 11 горожан — носителей мутации, ведущей к появлению болезни Лебера. Также установлено, что у франкоязычных канадцев чаще встречается мутация 14484С, которая является очень редкой в Великобритании и совсем не зарегистрирована в Финляндии [14, 20, 29]. Приведенные факты отражают высокую информативность генетических исследований для диагностики подобных состояний.
Время прогрессирования снижения зрения в большинстве случаев может быть охарактеризовано как острое или подострое со стабилизацией зрительных функций на низком уровне через дни, недели или месяцы. Среднее время стабилизации у большинства больных составляет около 4-6 недель. Начало развития симптомов заболевания характеризуется безболезненным ухудшением остроты центрального зрения на один глаз. Второй глаз вовлекается, как правило, несколькими неделями или месяцами позже. Однако существуют сообщения об одновременном начале и поражении обоих глаз. Конечная острота зрения в большинстве случаев не превышает сотые доли единицы. Цветовое зрение обычно поражается на ранних стадиях развития заболевания в значительной степени. Характерным является наличие дефекта зрительного поля с выявлением абсолютных или относительных скотом центральной/парацентральной локализации на белый и другие цвета. Использование цветового тестирования позволяет выявить нарушение зрительных функций на ранних стадиях, прежде чем возникает какое-либо доступное оценке снижение остроты зрения. Также необходимо отметить, что выявление аномального цветоощущения у асимптомных родственников не может достоверно предсказать вероятный риск развития данного заболевания. В большинстве случаев снижение зрения остается выраженным и постоянным [2, 28]. Однако, кроме заболевания классического течения, выделяют субклиническую форму, при которой отмечено медленное прогрессирование заболевания с незначительным снижением остроты зрения. Также описаны случаи спонтанного улучшения зрительных функций, иногда значительного уровня, которые могут иметь место спустя годы после манифестации. При офтальмологическом исследовании в течение острой фазы потери зрения, выявляется гиперемия сосков дисков зрительных нервов, дилатация и извитость сосудов мелкого и среднего размера, геморрагии и стушеванность краев дисков. Все вышеперечисленное интерпретируется чаще всего как признаки воспалительного процесса. Однако по данным флюоресцентной ангиографии проницаемость сосудов не изменена. В связи с этим необходимо отметить триаду патогномоничных изменений глазного дна у пациентов в острой фазе снижения зрения, что было выделено в исследованиях J.L. Smith et al. [33]: циркумпапиллярная телеангиоэктатическая микроангиопатия, набухание волокон нерва вокруг диска и отсутствие транссудации красителя при флюоресцентной ангиографии [32]. В динамике через несколько месяцев после начала заболевания телеангиоэктазии и псевдоэдема разрешаются, соски зрительных нервов начинают постепенно бледнеть и развивается простая атрофия зрительных нервов. Обычно бледнеет весь сосок, реже — только височная его часть, в папилломакулярной области, что может быть единственным оставшимся признаком по истечении острой фазы заболевания. В некоторых случаях могут встречаться признаки капиллярной микроангиопатии перипапиллярной области, в том числе и у асимптомных родственников, что может рассматриваться как диагностический маркер заболевания. У большинства пациентов с болезнью Лебера нарушение зрительных функций является наиболее значимым клиническим признаком заболевания [28, 29]. У мужчин риск значительного нарушения зрительных функций составляет от 20 до 83 %, а у женщин — 4-32 % [2]. В немногих описанных патологоанатомических исследованиях было установлено, что на всем протяжении зрительного нерва от глазного яблока до хиазмы резко выражен процесс атрофии нервных волокон с распадом мякотных влагалищ, разрастанием глии и изменениями мелких разветвлений соединительнотканных перекладин при полном отсутствии воспалительных изменений. Важным морфологическим признаком в случае болезни Лебера может быть набухание зрительных оболочек зрительных нервов, что отсутствует при атрофиях зрительных нервов другой этиологии [9]. Необходимо отметить, что в ряде родословных было установлено присутствие так называемых ассоциированных признаков. В родословных описаны больные, имеющие клинические признаки болезни Лебера в сочетании с тяжелыми неврологическими аномалиями, включающими случаи генерализованной дистонии, мягкой мозжечковой атаксии, дистальной сенсорной невропатии, спастического парапареза, тремора, судорог, мигрени, миоклонии, паркинсонизма и психических расстройств. Ранее существовало представление, что причиной болезни Лебера является оптико-хиазмальный арахноидит, по поводу чего проводилось хирургическое лечение в виде рассечения хиазмальных спаек. Однако подобные взгляды были пересмотрены и являются необоснованными, а изменения паутинной оболочки, обнаруживаемые в некоторых случаях во время операций, расценивают как вторичные [2]. У пациентов с болезнью Лебера неврологические нарушения выявляются до 59 % [24]. Одним из распространенных симптомов является тремор. По данным ряда авторов, он встречается в 20 % случаев. Отмечено, что тремор может быть и единственным проявлением носительства мутации 11778А. В литературе есть указания о возможности развития неврологических нарушений без патологии зрения у женщин — носительниц мутаций. Следует отметить, что болезнь Лебера достаточно часто приходится дифференцировать в клинике неврологии с рассеянным склерозом. При этом также описаны аналогичные наблюдающимся при рассеянном склерозе изменения при магнитно-резонансной томографии. Отмечено, что данное сочетание является наиболее типичным для мутации 11778А, а среди больных с этой мутацией — для женщин [3, 25, 28]. Также в ряде родословных описаны сочетания болезни Лебера с семейными аномалиями скелета. Особенно типичным является наличие кифосколиоза при болезни Лебера, обусловленной мутацией 3460А [24]. Также необходимо упомянуть о том факте, что при многих митохондриальных болезнях может наблюдаться сахарный диабет, что связано с мутациями как митохондриальной ДНК (синдромы Кeрнса — Сейра, MELAS), так и ядерной ДНК (атаксия Фридрейха, семейный шейный липоматоз). Однако сочетание болезни Лебера и сахарного диабета является крайне редким. Также не описана при наличии болезни Лебера и другая эндокринная патология [7, 11].
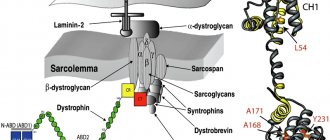
При анализе значимости вспомогательных диагностических методик было установлено, что при выполнении магнитно-резонансной томографии могут выявляться неспецифические повреждения зрительного нерва, а проведение таких дополнительных исследований, как электроэнцефалография, компьютерная томография и спинномозговая пункция, являются непоказательными [16, 33]. При проведении мышечной биопсии не отмечаются какие-либо характерные изменения, однако, по данным ряда авторов, выявлены увеличение размеров субсарколеммальных митохондрий без нарушений их структуры и незначительное варьирование размеров мышечных волокон, что предполагает наличие неспецифического миопатического процесса [2, 31].
При электрокардиографическом исследовании (ЭКГ) в 9 % случаев были обнаружены нарушения проведения по типу синдрома Вольфа — Паркинсона — Уайта (WPW), что обусловлено наличием дополнительных аномальных путей проведения электрического импульса от предсердий к желудочкам — так называемых пучков Кента [21, 26]. Эти пучки могут располагаться в любом месте вокруг правого или левого атриовентрикулярного кольца. При этом возбуждение проводится от предсердий к желудочкам как по обычному пути — атриовентрикулярному узлу (АВ-узлу) и пучку Гиса, так и по дополнительному аномальному пучку Кента. При этом пучок Кента проводит электрические импульсы гораздо быстрее, чем атриовентрикулярный узел, поэтому возбуждение желудочков при синдроме WPW начинается почти сразу после деполяризации предсердий. Это приводит к резкому укорочению интервала P-Q менее 0,12 с, что является одним из важнейших признаков преждевременного возбуждения желудочков. Волна возбуждения, проведенная из предсердий к желудочкам по дополнительному пучку Кента, медленно распространяется необычным путем по базальной части желудочка, что способствует появлению на ЭКГ дополнительной волны возбуждения желудочков — Д-волны, при столкновении которой с основной волной деполяризации (распространяющейся по АВ-узлу и пучку Гиса) возникает деформированный и уширенный комплекс QRS, что также является важным признаком синдрома WPW. Также отмечены такие нарушения сердечной проводимости, как синдром Клерка — Леви — Кристеско (CLC) c наличием дополнительного аномального пути проведения электрического импульса между предсердиями и пучком Гиса — пучок Джеймса. Этот пучок как бы шунтирует атриовентрикулярный узел, приводя к ускоренному возбуждению желудочков. В отличие от синдрома WPW волна возбуждения при синдроме CLC распространяется по желудочкам обычным путем (пучок Гиса, его ветви и волокна Пуркинье). Поэтому сам комплекс QRS не деформирован и не уширен, а для самого синдрома CLC имеет место укорочение интервала P-Q менее 0,12 с и обычно узкие, нормальной формы комплексы QRS без Д-волны. Учитывая данные особенности нарушений проводимости, у больных могут отмечаться приступы пароксизмальной суправентрикулярной тахикардии или мерцательной аритмии [3, 25, 27].
Следует отметить универсальность митохондриальной системы синтеза АТФ и высокую чувствительность тканей к внутриклеточному дефициту энергии, что определяет поражение различных органов и систем при патогенных мутациях митохондриальной ДНК, в частности при болезни Лебера. Было отмечено, что биохимический метод оценки респирации митохондрий в культуре лимфобластов и трансмитохондриальных цитоплазматических гибридов позволяет обнаружить дефект системы окислительного фосфорилирования на функциональном уровне [2]. Также установлено, что различные органы и ткани характеризуются разной степенью зависимости от активности системы окислительного фосфорилирования митохондрий. В порядке убывания наибольшей зависимостью обладают центральная нервная система (включая и орган зрения), миокард, скелетные мышцы, почки, эндокринные органы и печень, что обусловливает более частое их поражение при патогенных мутациях митохондриальной ДНК [1, 31].
С целью диагностики и дифференциальной диагностики следует отметить, что атрофия зрительных нервов является одним из признаков ряда наследственных патологических состояний, которые обусловлены отклонениями нормального функционирования дыхательной цепи митохондрий. В связи с этим можно привести такие нозологические формы, как NARP — неврологические расстройства в сочетании с пигментной ретинопатией, MERRF — миоклоническая эпилепсия с синдромом «красных рваных волокон» и CPEO — хроническая прогрессирующая наружная офтальмоплегия. При этих заболеваниях, также как и при болезни Лебера, может присутствовать атрофия зрительных нервов [1, 23, 31]. Поэтому для подтверждения диагноза необходимо проведение молекулярно-генетического исследования с обнаружением патогенных мутаций митохондриальной ДНК, что является единственным общепринятым признаком заболевания и позволяет достоверно установить наличие болезни Лебера даже в отсутствие характерного семейного анамнеза.
Необходимо отметить, что восстановление зрения вариабельно и зависит от выявленной мутации. Наилучший прогноз характерен для 14484-й мутации (50 % больных) [18], улучшение зрения отмечается менее чем у 5 % больных с 11778-й мутацией [22, 26], частота выздоровления у больных с 15275-й мутацией составляет 25 % [15], а больные моложе 15 лет имеют лучший прогноз независимо от типа мутации.
Таким образом, болезнь Лебера является серьезной проблемой современной медицины. Это обусловлено тем важным фактом, что подходы к лечению митохондриальной патологии в настоящее время за рубежом находятся в стадии разработки, наличием как моносимптомного характера течения этого заболевания, так и в некоторых случаях многосистемностью поражения и многосимптомностью клинической картины, что может вызывать трудности в диагностике и дифференциальной диагностике. Поэтому следует еще раз подчеркнуть значение молекулярно-генетических методов исследования, помогающих в постановке диагноза. Также неоднозначной и трудной является оценка хирургического лечения болезни Лебера, что связано с высоким риском осложнений. Следует отметить, что предпринимались неоднократные попытки воздействия на увеличение продукции энергии в митохондриях с использованием природных препаратов в виде кофермента Q10, витаминов K1, K3, С, В2 и сукцината, однако их использование в лечении болезни Лебера не было успешным. Существуют отдельные сообщения об эффективности применения идебенона (производное Q10) при его длительном использовании у пациентов с болезнью Лебера [8]. Это определяет необходимость и целесообразность поиска новых средств и подходов для лечения данной патологии с реализацией клинической эффективности при их использовании. Своевременная диагностика позволяет избежать ненужных обследований для реализации клинической эффективности проводимого лечения и прогноза течения заболевания.
Литература 1. Евтушенко С.К. Метаболический (митохондриальный) инсульт у детей // Международный неврологический журнал. — 2008. — № 2. — С. 10-15. 2. Жаданов С.И. Наследственная зрительная атрофия Лебера. Новые перспективы исследований // Новое в офтальмологии — 2001. — № 2. — С. 28-37. 3. Мурашко В.В., Струтынский А.В. Электрокардиография. — Москва: МЕДпресс, 1999. — С. 190-193. 4. Руденская Г.Е., Захарова Е.Ю., Адарчева Л.С., Михайлова Е.Н., Карлова И.З. Наследственная атрофия зрительных нервов Лебера: неврологические и другие внеглазные проявления // Журнал неврологии и психиатрии. — 2004. — № 2. — С. 40. 5. Brown M.D., Torroni A., Reckord C.L., Wallace D.C. Phylogenetic analysis of caucacian 11778-positive and 11778-negative Leber’s hereditary optic neuropathy patients indicates multiple indepent occurrences of the common primary mitochondrial DNA mutations // Hum. Mutat. — 1995. — Vol. 6. — P. 311-325. 6. Chalmers R.M., Schapira A.H. Clinical, biochemical and molecular genetic features of Leber’s hereditary optic neuropathy // Biochim. Biophys. Acta. — 1999. — Vol. 1410. — P. 147-58. 7. Chinnery P., Johnson M., Wardell T. The epidemiology of pathogenic mitochondrial DNA mutations // Ann. Neurol. — 2000. — Vol. 48. — P. 188-193. 8. Cole A., Dutton C. Leber’s hereditary optic neuropathy and maturity onset diabetes mellitus: is there a metabolic association // Br. J. Ophthalmol. — 2000. — Vol. 84. — P. 439-440. 9. Cortelli P., Montagna P., Pierangeli G. Clinical and brain bioenergetics improvement with idebenone in a patients with Leber’s hereditary optic neuropathy: a clinical and 31P-MRS study // J. Neurol. Sci. — 1997. — Vol. 148 (1). — P. 25-31. 10. De Gottrau P., Buchi E.R., Daicker B. Distended optic nerve sheaths in Leber’s hereditary optic neuropathy // J. Clin. Neuroophthalmol. — 1992. — Vol. 12 (2). — P. 89-93. 11. Dotti M., Plewnia K., Cardaoli E. A case of ethambutol-induced optic neuropathy harbouring the primary LHON mutation at 11778 // J. Neurol. — 1998. — Vol. 245. — P. 302-303. 12. DuBois L., Feldon S. Evidence for a metabolic trigger for Leber’s hereditary optic neuropathy. A case report // J. Clin. Ophthalmol. — 1992. — Vol. 12. — P. 15-16. 13. Feng X., Pu W., Gao D. Diagnostic and differential diagnostic potential of mitochondrial DNA assessment in patients with Leber’s hereditary optic neuropathy // Chung Hua Yen Ko Tsa Chih. — 2001. — Vol. 37. — P. 174-177. 14. Howell N. LHON and other optic nerve atrophies: the mitochondrial connection // Ophthalmol. — 2003. — Vol. 37. — P. 94-108. 15. Huoponen K. Leber hereditary optic neuropathy: clinical and molecular genetic findings // Neurogenetics. — 2001. — Vol. 3. — P. 119-125. 16. Johns D.R., Smith K.H., Savino P.J. Leber’s hereditary optic neuropathy: clinical manifestations of the 15257 mutation // Ophthalmology. — 1993. — Vol. 100. — P. 981-986. 17. Kermode A.G., Moseley I.F., Kendall B.E. Magnetic resonance imaging in Leber’s optic neuropathy // J. Neurol. Neurosurg. Psychiatry. — 1989. — Vol. 52. — P. 671-674. 18. Mackey D.A., Oostra R.J., Rosenberg T. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy // Am. J. Hum. Genet. — 1996. — Vol. 59. — P. 4815. 19. Mackey D.A., Howell N. A variant of Leber hereditary optic neuropathy characterized by recovery of vision and by an unusual mitochondrial genetic etiology // Am. J. Hum. Genet. — 1992. — Vol. 51. — P. 1218-1228. 20. Mackey D.A., Buttery R. Leber’s hereditary optic neuropathy in Australia // Aust. New Zeal. J. Ophthalmol. — 1992. — Vol. 20. — P. 177-184. 21. Macmillan C., Kirkham T., Fu K. Pedigree analysis of French Z Canadian Families with T14484C Leber’s hereditary optic neuropathy // Neurology. — 1988. — Vol. 50. — P. 417-422. 22. Mashima Y., Kigasawa K., Hasegawa H. High incidence of pre-excitation syndrome in Japenese families with Leber’s hereditary optic neuropathy // Clin. Genet. — 1996. — Vol. 50 (6). — P. 535-537. 23. Newman N.J., Lott M.T., Wallace D.C. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation // Am. J. Ophthaimol. — 1991. — Vol. 111. — P. 750-762. 24. Newman N.J. Mitochondrial disease and the eye. Neuro-ophthalmology in systemic disease. — 1992. — Р. 3. 25. Nikoskelainen E.K., Marttila R.J., Huoponen K., Juvonen V. Leber’s «plus»: neurological abnormalities in patients with Leber’s hereditary optic neuropathy // J. Neurol. Neurosurg. Psychiatry. — 1995. — Vol. 59 (2). — P. 160-164. 26. Nikoskelainen E.K., Savontaus M.-L., Huoponen K. Pre-excitation syndrome in Leber’s hereditary optic neuroretinopathy // Lancet. — 1994. — Vol. 344 (8296). — P. 857-858. 27. Oostra R.J., Bolhuis P.A., Wijburg F.A. Leber’s hereditary optic neuropathy: correlations between mitochondrial genotype and visual outcome // J. Med. Genet. — 1994. — Vol. 31. — P. 280-286. 28. Ortiz R.G., Newman N.J., Manoukian S.V. Optic disk cupping and electrographic abnormalities in an American pedigree with Leber’s hereditary optic neuropathy // Am. J. Ophthalmol. — 1992. — Vol. 113. — P. 561-566. 29. Riordan-Eva P., Sanders M.D., Govan G.G. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation // Brain. — 1995. — Vol. 118 (2). — P. 319-337. 30. Scott C.N. Oliver, Jeffrey L. Bennett. Genetic disorders and the optic nerve: a clinical survey // Ophthalmology Clinics of North America. — 2004. — Vol. 17(3). — P. 435-445. 31. Shaikh S., Ta C., Basham A., Mansour S. Leber hereditary optic neuropathy associated with antiretroviral therapy for human immunodeficiency virus infection // Am. J. Ophthalmol. — 2001. — Vol. 131. — P. 143-145. 32. Shoffner J.M., Wallace D.C. Oxidative phosphor rylation diseases. Disorders of two genomes // Adv. Hum. Genet. — 1990. — Vol. 19. — P. 267. 33. Smith J.L., Hoyt W.F., Susac J.O. Ocular fundus in acute Leber hereditary optic neuropathy // Arch. Ophthalmol. — 1973. — Vol. 90. — P. 349. 34. Vaphiades M.S., Newman N.J. Optic nerve enhancement on orbital magnetic resonance imaging in Leber’s hereditary optic neuropathy // J. Neuroophthalmol. — 1999. — Vol. 19. — P. 238-239. 35. Wallace D.C., Singh G., Lott M.T., Hodge J.A. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy // Science. — 1988. — Vol. 242. — P. 1427-1430. 36. Yeates F.M. Causes of binocular legal blindness in an Australian metropolitan community // Aust. J. Ophthalmol. — 1983. — Vol. 11. — P. 321-323.
Причины наследственной оптической нейропатии Лебера
Причины нейропатии Лебера, при которой атрофируется зрительный нерв, — генетические. Болезнь, обусловленная специфической мутацией генов митохондрий, передается женщинами своему потомству. Мужчины и женщины наследуют патологию от матери. Почему так происходит? Огромное количество ДНК находится в ядре клеток и лишь небольшая его часть — в митохондриях. Гены ядра наследуются и от матери, и от отца, а вот гены митохондрии — только от матери. Мужчина, унаследовавший мутацию, не передаст ее своим потомкам.
У значительной части людей, которые являются носителем мутированного гена, болезнь не станет явной. Более 85% женщин и 50% мужчин, которые являются носителем гена синдрома LHON, болезнь не затронет. Причины, влияющие на прогрессирование заболевания, не ясны, но достоверно известно, что спровоцировать болезнь может неблагоприятная экологическая обстановка, стрессы, инфекции, токсическое воздействие табака и алкоголя.
Диагностика оптической нейропатии Лебера
Диагностика болезни осложнена схожестью ее симптомов с ишемической невропатией или с определенными патологиями — например, с рассеянным склерозом. Сложно отследить и анамнез, так как не у всех носителей мутировавшего гена развивается нейропатия Лебера. Расширенное офтальмологическое обследование может только установить наличие патологии, а вот для определения причины, понадобится генетическая диагностика.
Особенности диагностики наследственной оптической нейропатии Лебера:
- общее клиническое обследование;
- анализ митохондриальной ДНК;
- исследование полей зрения;
- осмотр глазного дна;
- когерентная томография и электроретинография (необходимы, чтобы исключить патологии сетчатки).
Специалистам рекомендуется проводить дифференциальную диагностику синдрома LHON, сравнивая симптоматику и данные обследования с другими заболеваниями, поражающими глазной нерв. Например, с ишемической, токсической невропатией. Полное офтальмологическое обследование показывает, что диск зрительного нерва воспаленный, также заметны телеангиэктазии сосудов (расширение мелких капилляров).
Как проявляет себя заболевание?
Заподозрить атрофию оптического нерва Лебера можно по нескольким характерным признакам:
- У человека резко (иногда — постепенно в течение пары лет) снижается зрительная острота. Падение зрения может сразу идти как двусторонний процесс, либо сначала зрение ухудшается на одном, а потом — на втором глазу.
- В центре зрительного поля могут появиться так называемые скотомы — темные «слепые» пятна.
- Для ранней стадии заболевания характерно покраснение диска оптического нерва. Спустя пару недель — его височная половина бледнеет. Данный симптом можно заметить только при офтальмологическом обследовании.
- Примерно в половине случаев, наряду с признаками поражения глаз, могут наблюдаться неврологические проявления болезни — тремор, головокружение, ухудшение рефлексов, головная боль, ярко выраженный мышечный тонус.
Наследственная оптическая нейропатия Лебера — можно ли вылечить болезнь?
Пока наследственная нейропатия оптическая считается неизлечимым заболеванием, но учеными ведется постоянный поиск новых путей для терапии подобной патологии. Благодаря методам генной терапии можно существенно замедлить прогрессирование болезней и улучшить качество жизни пациентов.
Стремительное прогрессирование болезни Лебера приводит к быстрой потере зрения людьми, которые всю свою жизнь видели нормально и не имели проблем со зрением. Неутешительный диагноз, а также информация о том, что болезнь пока не лечится, серьезно сказываются на самочувствии таких пациентов, которые не были готовы к инвалидности. Многие из существующих методик лечения патологии оказались малоэффективными, в том числе и хирургическое вмешательство.
К счастью, ученые, занимающиеся редкими патологиями, прикладывают немало усилий к тому, чтобы найти средство излечения. Особую надежду специалисты возлагают на генную терапию, и у многих исследовательских групп уже есть неплохие результаты. Иногда препятствием к продолжению экспериментов является их этическая сторона, так как дальнейшие исследования предполагают использование разработанной методики на людях. Есть ли успехи хотя бы с использованием экспериментальных моделей? Да, генетикам уже удалось найти путь к решению проблемы.
Так, группа ученых из Майами, используя экспериментальные модели, доказала, что мутировавшие гены можно безопасно заменить здоровыми, это предотвратит ухудшение питания клеток зрительного нерва. Для исправления генетического дефекта в митохондрии необходимо ввести нормальную ДНК — это позволит исправить нарушение и восстановить зрительную функцию. Ученые сообщают, что такой подход будет эффективным и в отношении других заболеваний, вызванных митохондриальными мутациями, а также различных нарушений, связанных с процессами старения организма.
Причины
В большинстве случаев фиксируется передача заболевания аутосомно-рецессивным способом. Это означает, что патология зрения будет вызвана у ребёнка в том случае, если оба родителя будут иметь дефектный ген.
По причине того, что клетки пигментного эпителия и фоторецепторов у новорожденного не способны разделяться, как-раз и проявляется амавроз Лебера. На сегодняшний день данное заболевание считается гетерогенной группой нарушений, которые вызывают уничтожение колбочек и палочек.