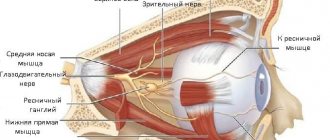
A sharp bilateral decrease in visual acuity may indicate the development of a dangerous pathology - Leber optic neuropathy. This disease most often affects young males. Let's consider the causes of pathology, symptoms and features of therapy in the article.
For the first time, cases of a disease in which visual acuity rapidly decreases due to atrophy of the optic nerve were described in 1871 by Theodor Leber. The scientist observed the development of pathology in young people from several related families, which indicated a hereditary cause of the disease. The mechanism of inheritance of the disease was carefully studied, so it was possible to prove that the pathology is transmitted through the maternal line mainly to men.
Until now, the nature of the disease has not been studied well enough to find effective treatments for such a disease. Currently, treatment of patients consists of maintenance therapy, but from time to time groups of scientists present new gene therapy techniques for clinical trials, which, of course, gives hope to people with a similar diagnosis.
Leber's optic neuropathy: symptoms, treatment of the disease
Any visual impairment that rapidly progresses must be carefully studied and comprehensively diagnosed. Leber's optic neuropathy is rarely accompanied by malaise characteristic of neurological diseases, and vision declines quickly, sometimes not even within a few months, but within two to three weeks. With such acuity, vision cannot be checked using Sivtsev’s table, because a person simply does not see the signs, so they use hardware diagnostics and the “counting fingers” method at a certain distance from the patient’s face. The peculiarity of the disease is that the optic nerve is affected due to a gene mutation. An excess amount of toxic oxygen molecules is produced, which negatively affects the condition of nerve cells and vision decreases.
Leber's optic neuropathy - symptoms and features of the disease:
- young age of patients - 18-35 years;
- the disease develops more often in men;
- rapid unilateral and then bilateral vision loss;
- visual acuity decreases over several weeks;
- violation of color perception of red and green colors;
- the pathology may be accompanied by symptoms inherent in mitochondrial diseases (convulsions, cardiac conduction disturbances).
With Leber optic nerve atrophy, most often the process of disease development begins in one eye, but there are often cases when two eyes are affected at once. Central vision is constantly decreasing. This is due to the gradual death of cells in the optic nerve, which transmits image information to the brain. Sometimes the disease may be accompanied by symptoms characteristic of neurological diseases. In this case, the sick person suffers from dystonia, tremor, and ataxia. Less often, at the beginning of the development of pathology, a person notices sparks, spots, bright flashes before the eyes.
Leber optic atrophy: clinical and genetic aspects (scientific review)
L.G. Kirillova, A.A. Shevchenko, L.Yu. Silaeva, State Institution “Institute of Pediatrics, Obstetrics and Gynecology of the Academy of Medical Sciences of Ukraine”, Kyiv
Summary
Modern views on the problem of mitochondrial disease - Leber optic atrophy, an assessment of various diagnostic and treatment methods, as well as the need for molecular genetic research to detect pathogenic mutations in mitochondrial DNA are presented. Timely diagnosis allows you to avoid unnecessary examinations to realize the clinical effectiveness of the treatment and prognosis of the course of the disease.
Keywords
Leber optic atrophy, mitochondrial disease, molecular genetic study.
In recent years, the classical clinic of many diseases in neuropediatrics has changed so much that an experienced clinician often mentally asks himself the question: “What is the clinic and the course of the pathological process that I am faced with?” Sometimes it is very difficult at a council of doctors to come to a consensus about a particular disease, and this difficulty in many cases can be explained by the fact that we, knowing about the existence of a number of mitochondrial diseases, do not have the opportunity to either confirm or exclude the alleged pathology without in-depth genetic research.
In practice, we have to deal with the situation when parents with children who progressively lose visual acuity with ophthalmological changes in the fundus turn to a pediatrician and then to a neurologist. In such cases, it is necessary first of all to exclude various types of inflammatory processes, brain tumors and hereditary degenerative diseases. Rarely do we think about Leber's mitochondrial optic atrophy. The reason for writing this article was two cases of illness in children who were in the pediatric psychoneurology department of the IPAG of the Academy of Medical Sciences of Ukraine over the past year (one of them was diagnosed with Leber’s congenital atrophy in Moscow and was genetically confirmed).
Leber optic atrophy has been known as a familial form of blindness since the late 19th century, when the German ophthalmologist Theodor Leber first described this pathology in 1871. This disease is manifested by a rapid bilateral decrease in vision with central scotomas, color vision disorder and is inherited in a recessive, sex-linked manner. Considering that the main substrate of the lesion is the optic nerve, and in many cases there are other neurological disorders and lesions, Leber's disease can be classified as a neurological disease [3, 29]. It should also be noted that Leber visual atrophy is a disease in which hereditary point mutations in mitochondrial DNA have been discovered. In 1988, DC Wallace et al. It was found that Leber optic nerve atrophy is associated with a substitution of the 11778th nucleotide of the mitochondrial gene encoding the 4th subunit of complex I of the respiratory chain. Subsequently, other mutations leading to this disease were discovered. In most cases, they affect mitochondrial genes encoding proteins that are involved in electron transfer in the respiratory chain with disruption of the phosphorylating function of mitochondria. In 95% of cases, three mutations are detected in mitochondrial DNA at positions 11778, 3460 and 14484 [4, 15, 17]. Mutation 11778A (substitution of guanine/adenyl bases at position 11778 of mitochondrial DNA, leading to the replacement of arginine by histidine in the 340th codon of the NADH dehydrogenase subunit 4 gene) occurs in 69% of cases in almost all population groups. Mutation 14484C (replacement of thiamine/cytosine bases at position 14484C of mitochondrial DNA, leading to the replacement of methionine with valine in the NADH dehydrogenase subunit 6 gene) occurs in 14% of cases, concentrating in people from the Netherlands and England, and mutation 3460A (replacement of guanine bases /adenine at position 3460 of mitochondrial DNA, leading to the replacement of alanine with threonine in the 52nd codon of the NADH dehydrogenase subunit 1 gene) - in approximately 13% of cases. The 15275 mutation was found in a minority of families. Mitochondrial mutations leading to Leber's disease are transmitted from the mother to all children, but the disease develops predominantly in sons. It has been established that the ratio of affected men and women, respectively, is 5: 1, and the average age of manifestation is from 23 to 26 years (the earliest onset is 4 years and the latest is 86 years), congenital forms are observed mainly in children. It was also found that this pathology most often occurs in residents of Northern Europe or the Japanese, and there are interpopulation differences in the ratio of primary mutations. For example, in Asia the proportion of mutation 11778A is higher than in Western countries [13, 29, 35]. According to epidemiological data, the frequency of Leber's disease in north-east England is 3.3 per 105 population, the carriage rate is 8.9 per 105, and in Finland its prevalence is about 1: 50,000 [6, 14]. In Australia, among all registered blind people, patients with Leber optic atrophy account for 0.4-2% [19]. Among the external factors that increase the penetrance of the mutant gene and provoke the manifestation of Leber's disease are alcohol consumption and smoking. At the same time, the effect of smoking is associated with the effect of cyanide, carbon monoxide and other toxins of tobacco smoke on the processes of oxidative phosphorylation. There are also individual reports of provoking external factors in the form of traumatic brain injuries, psycho-emotional stress, androgenic drugs, the anti-tuberculosis drug ethambutol and antiviral drugs used for AIDS [10, 12, 22, 30]. The described clinical cases of Leber's disease are represented by both extensive pedigrees with a large number of affected individuals, and sporadic cases of optic atrophy in the absence of indications of a characteristic family history. Of interest is a study conducted by Canadian geneticists from the universities of Quebec and Montreal, which made it possible to determine the origin of Leber's disease in Quebec. They were able to identify the girl who passed this rare genetic disease on to her descendants, the residents of Quebec. She turned out to be one of the 700 orphan girls whom the French king Louis XIV in 1663-1673 sent to the Belle Province (as Quebec was then called) to improve and radically change the demographic situation there. Using an extensive database of all Quebecers born after 1800, the genealogy of 11 city residents who carried the mutation leading to the appearance of Leber's disease was established. It has also been found that French-speaking Canadians are more likely to have the 14484C mutation, which is very rare in the UK and has not been registered at all in Finland [14, 20, 29]. The above facts reflect the high informativeness of genetic studies for diagnosing such conditions.
The progression of vision loss in most cases can be characterized as acute or subacute, with visual function stabilizing at a low level after days, weeks or months. The average stabilization time for most patients is about 4-6 weeks. The onset of symptoms of the disease is characterized by a painless deterioration in the acuity of central vision in one eye. The second eye is usually involved several weeks or months later. However, there are reports of simultaneous onset and involvement of both eyes. The final visual acuity in most cases does not exceed hundredths of one. Color vision is usually affected to a significant extent in the early stages of the disease. Characteristic is the presence of a defect in the visual field with the identification of absolute or relative scotomas of central/paracentral localization in white and other colors. The use of color testing can detect visual impairment in the early stages, before any measurable loss of visual acuity occurs. It should also be noted that identifying abnormal color perception in asymptomatic relatives cannot reliably predict the likely risk of developing this disease. In most cases, vision loss remains pronounced and permanent [2, 28]. However, in addition to the classical disease, there is a subclinical form, in which slow progression of the disease is noted with a slight decrease in visual acuity. Cases of spontaneous improvement in visual function, sometimes to a significant level, which can occur years after manifestation, have also been described. An ophthalmological examination during the acute phase of vision loss reveals hyperemia of the nipples of the optic discs, dilatation and tortuosity of small and medium-sized vessels, hemorrhages and blurring of the edges of the discs. All of the above is most often interpreted as signs of an inflammatory process. However, according to fluorescein angiography, vascular permeability is not changed. In this regard, it is necessary to note the triad of pathognomonic changes in the fundus of the eye in patients in the acute phase of vision loss, which was identified in the studies of JL Smith et al. [33]: circumpapillary telangiectatic microangiopathy, swelling of nerve fibers around the disc and absence of dye extravasation on fluorescein angiography [32]. Over time, several months after the onset of the disease, telangiectasia and pseudoedema resolve, the papillae of the optic nerves begin to gradually turn pale, and simple atrophy of the optic nerves develops. Usually the entire nipple turns pale, less often - only its temporal part, in the papillomacular region, which may be the only remaining sign after the acute phase of the disease. In some cases, signs of capillary microangiopathy of the peripapillary region may occur, including in asymptomatic relatives, which can be considered as a diagnostic marker of the disease. In most patients with Leber disease, visual impairment is the most significant clinical sign of the disease [28, 29]. In men, the risk of significant visual impairment ranges from 20 to 83%, and in women - 4-32% [2]. In the few described pathoanatomical studies, it was found that throughout the entire length of the optic nerve from the eyeball to the chiasm, the process of atrophy of nerve fibers with the disintegration of the pulpy sheaths, the proliferation of glia and changes in the small branches of the connective tissue crossbars in the complete absence of inflammatory changes is clearly expressed. An important morphological sign in the case of Leber's disease may be swelling of the optic membranes of the optic nerves, which is absent in optic nerve atrophies of other etiologies [9]. It should be noted that in a number of pedigrees the presence of so-called associated characteristics was established. Pedigrees describe patients with clinical features of Leber disease in combination with severe neurological abnormalities, including cases of generalized dystonia, mild cerebellar ataxia, distal sensory neuropathy, spastic paraparesis, tremor, seizures, migraine, myoclonus, parkinsonism and mental disorders. Previously, there was an idea that the cause of Leber's disease was optico-chiasmatic arachnoiditis, for which surgical treatment was carried out in the form of dissection of the chiasmatic adhesions. However, such views have been revised and are unfounded, and changes in the arachnoid membrane, detected in some cases during operations, are regarded as secondary [2]. In patients with Leber's disease, neurological disorders are detected in up to 59% [24]. One common symptom is tremors. According to a number of authors, it occurs in 20% of cases. It was noted that tremor may be the only manifestation of carriage of the 11778A mutation. There are indications in the literature about the possibility of developing neurological disorders without visual pathology in women who are carriers of mutations. It should be noted that Leber's disease quite often has to be differentiated in a neurology clinic from multiple sclerosis. At the same time, changes similar to those observed in multiple sclerosis on magnetic resonance imaging are also described. It has been noted that this combination is most typical for the 11778A mutation, and among patients with this mutation - for women [3, 25, 28]. Also, a number of pedigrees describe combinations of Leber's disease with familial skeletal anomalies. Particularly typical is the presence of kyphoscoliosis in Leber disease, caused by the 3460A mutation [24]. It is also necessary to mention the fact that diabetes mellitus can be observed in many mitochondrial diseases, which is associated with mutations in both mitochondrial DNA (Cairns-Sayre syndromes, MELAS) and nuclear DNA (Friedreich's ataxia, familial cervical lipomatosis). However, the combination of Leber's disease and diabetes mellitus is extremely rare. Other endocrine pathologies have also not been described in the presence of Leber’s disease [7, 11].
When analyzing the significance of auxiliary diagnostic techniques, it was found that when performing magnetic resonance imaging, nonspecific damage to the optic nerve can be detected, and additional studies such as electroencephalography, computed tomography and spinal puncture are non-indicative [16, 33]. When performing a muscle biopsy, no characteristic changes are noted, however, according to a number of authors, an increase in the size of subsarcolemmal mitochondria without disturbances in their structure and a slight variation in the size of muscle fibers were detected, which suggests the presence of a nonspecific myopathic process [2, 31].
An electrocardiographic study (ECG) revealed conduction disturbances of the Wolf-Parkinson-White (WPW) syndrome type in 9% of cases, which is due to the presence of additional abnormal pathways for conducting electrical impulses from the atria to the ventricles - the so-called bundles of Kent [21, 26]. These bundles can be located anywhere around the right or left atrioventricular ring. In this case, excitation is carried out from the atria to the ventricles both along the usual path - the atrioventricular node (AV node) and the His bundle, and through the additional anomalous bundle of Kent. At the same time, the Kent bundle conducts electrical impulses much faster than the atrioventricular node, so excitation of the ventricles in WPW syndrome begins almost immediately after atrial depolarization. This leads to a sharp shortening of the PQ interval to less than 0.12 s, which is one of the most important signs of premature ventricular excitation. The excitation wave, conducted from the atria to the ventricles along the additional Kent bundle, slowly spreads in an unusual way along the basal part of the ventricle, which contributes to the appearance on the ECG of an additional wave of ventricular excitation - the D-wave, when it collides with the main depolarization wave (propagating along the AV node and His bundle), a deformed and widened QRS complex occurs, which is also an important sign of WPW syndrome. Also noted are cardiac conduction disorders such as Clerk-Lewy-Christesco syndrome (CLC) with the presence of an additional abnormal path of electrical impulse between the atria and the His bundle - the James bundle. This bundle, as it were, shunts the atrioventricular node, leading to accelerated excitation of the ventricles. Unlike WPW syndrome, the excitation wave in CLC syndrome propagates through the ventricles in the usual way (the His bundle, its branches and Purkinje fibers). Therefore, the QRS complex itself is not deformed or widened, and for the CLC syndrome itself, there is a shortening of the PQ interval of less than 0.12 s and usually narrow, normal-shaped QRS complexes without a D-wave. Considering these features of conduction disorders, patients may experience attacks of paroxysmal supraventricular tachycardia or atrial fibrillation [3, 25, 27].
It should be noted the universality of the mitochondrial ATP synthesis system and the high sensitivity of tissues to intracellular energy deficiency, which determines the damage to various organs and systems due to pathogenic mutations of mitochondrial DNA, in particular in Leber's disease. It was noted that the biochemical method for assessing mitochondrial respiration in the culture of lymphoblasts and transmitochondrial cytoplasmic hybrids makes it possible to detect a defect in the oxidative phosphorylation system at the functional level [2]. It has also been established that different organs and tissues are characterized by varying degrees of dependence on the activity of the mitochondrial oxidative phosphorylation system. In descending order, the central nervous system (including the organ of vision), myocardium, skeletal muscles, kidneys, endocrine organs and liver have the greatest dependence, which causes more frequent damage to them due to pathogenic mutations of mitochondrial DNA [1, 31].
For the purpose of diagnosis and differential diagnosis, it should be noted that optic nerve atrophy is one of the signs of a number of hereditary pathological conditions that are caused by deviations in the normal functioning of the mitochondrial respiratory chain. In this regard, we can cite such nosological forms as NARP - neurological disorders in combination with pigmentary retinopathy, MERRF - myoclonic epilepsy with red ragged fiber syndrome and CPEO - chronic progressive external ophthalmoplegia. In these diseases, as well as in Leber's disease, atrophy of the optic nerves may be present [1, 23, 31]. Therefore, to confirm the diagnosis, it is necessary to conduct a molecular genetic study with the detection of pathogenic mutations in mitochondrial DNA, which is the only generally accepted sign of the disease and allows one to reliably establish the presence of Leber’s disease even in the absence of a characteristic family history.
It should be noted that vision recovery is variable and depends on the identified mutation. The best prognosis is typical for the 14484th mutation (50% of patients) [18], improvement in vision is observed in less than 5% of patients with the 11778th mutation [22, 26], the recovery rate in patients with the 15275th mutation is 25% [ 15], and patients under 15 years of age have a better prognosis regardless of the type of mutation.
Thus, Leber's disease is a serious problem in modern medicine. This is due to the important fact that approaches to the treatment of mitochondrial pathology are currently under development abroad, the presence of both the monosymptomatic nature of the course of this disease, and in some cases the multisystem nature of the lesion and the multisymptomatic clinical picture, which can cause difficulties in diagnosis and differential diagnostics. Therefore, it is necessary to once again emphasize the importance of molecular genetic research methods that help in making a diagnosis. Also controversial and difficult is the assessment of surgical treatment of Leber's disease, which is associated with a high risk of complications. It should be noted that repeated attempts have been made to increase energy production in mitochondria using natural preparations in the form of coenzyme Q10, vitamins K1, K3, C, B2 and succinate, but their use in the treatment of Leber's disease was not successful. There are isolated reports of the effectiveness of idebenone (Q10 derivative) with long-term use in patients with Leber's disease [8]. This determines the need and feasibility of searching for new means and approaches to treat this pathology with the realization of clinical effectiveness when using them. Timely diagnosis allows you to avoid unnecessary examinations to realize the clinical effectiveness of the treatment and prognosis of the course of the disease.
Literature 1. Evtushenko S.K. Metabolic (mitochondrial) stroke in children // International Neurological Journal. - 2008. - No. 2. - P. 10-15. 2. Zhadanov S.I. Leber's hereditary visual atrophy. New prospects for research // New in ophthalmology - 2001. - No. 2. - P. 28-37. 3. Murashko V.V., Strutynsky A.V. Electrocardiography. - Moscow: MEDpress, 1999. - P. 190-193. 4. Rudenskaya G.E., Zakharova E.Yu., Adarcheva L.S., Mikhailova E.N., Karlova I.Z. Leber's hereditary optic atrophy: neurological and other extraocular manifestations // Journal of Neurology and Psychiatry. - 2004. - No. 2. - P. 40. 5. Brown MD, Torroni A., Reckord CL, Wallace DC Phylogenetic analysis of caucacian 11778-positive and 11778-negative Leber's hereditary optic neuropathy patients indicates multiple indepent occurrences of the common primary mitochondrial DNA mutations // Hum. Mutat. - 1995. - Vol. 6. - P. 311-325. 6. Chalmers RM, Schapira AH Clinical, biochemical and molecular genetic features of Leber's hereditary optic neuropathy // Biochim. Biophys. Acta. - 1999. - Vol. 1410. - P. 147-58. 7. Chinnery P., Johnson M., Wardell T. The epidemiology of pathogenic mitochondrial DNA mutations // Ann. Neurol. - 2000. - Vol. 48. - P. 188-193. 8. Cole A., Dutton C. Leber's hereditary optic neuropathy and maturity onset diabetes mellitus: there is a metabolic association // Br. J. Ophthalmol. - 2000. - Vol. 84. - P. 439-440. 9. Cortelli P., Montagna P., Pierangeli G. Clinical and brain bioenergetics improvement with idebenone in a patient with Leber's hereditary optic neuropathy: a clinical and 31P-MRS study // J. Neurol. Sci. - 1997. - Vol. 148(1). — P. 25-31. 10. De Gottrau P., Buchi ER, Daicker B. Distended optic nerve sheaths in Leber's hereditary optic neuropathy // J. Clin. Neuroophthalmol. - 1992. - Vol. 12(2). — P. 89-93. 11. Dotti M., Plewnia K., Cardaoli E. A case of ethambutol-induced optic neuropathy harboring the primary LHON mutation at 11778 // J. Neurol. - 1998. - Vol. 245. - P. 302-303. 12. DuBois L., Feldon S. Evidence for a metabolic trigger for Leber's hereditary optic neuropathy. A case report // J. Clin. Ophthalmol. - 1992. - Vol. 12. - P. 15-16. 13. Feng X., Pu W., Gao D. Diagnostic and differential diagnostic potential of mitochondrial DNA assessment in patients with Leber's hereditary optic neuropathy // Chung Hua Yen Ko Tsa Chih. - 2001. - Vol. 37. - P. 174-177. 14. Howell N. LHON and other optic nerve atrophies: the mitochondrial connection // Ophthalmol. - 2003. - Vol. 37. - P. 94-108. 15. Huoponen K. Leber hereditary optic neuropathy: clinical and molecular genetic findings // Neurogenetics. - 2001. - Vol. 3. - P. 119-125. 16. Johns DR, Smith KH, Savino PJ Leber's hereditary optic neuropathy: clinical manifestations of the 15257 mutation // Ophthalmology. - 1993. - Vol. 100. - P. 981-986. 17. Kermode AG, Moseley IF, Kendall BE Magnetic resonance imaging in Leber's optic neuropathy // J. Neurol. Neurosurg. Psychiatry. - 1989. - Vol. 52. - P. 671-674. 18. Mackey DA, Oostra RJ, Rosenberg T. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy // Am. J.Hum. Genet. - 1996. - Vol. 59. - P. 4815. 19. Mackey DA, Howell N. A variant of Leber hereditary optic neuropathy characterized by recovery of vision and by an unusual mitochondrial genetic etiology // Am. J.Hum. Genet. - 1992. - Vol. 51. - P. 1218-1228. 20. Mackey DA, Buttery R. Leber's hereditary optic neuropathy in Australia // Aust. New Zeal. J. Ophthalmol. - 1992. - Vol. 20. - P. 177-184. 21. Macmillan C., Kirkham T., Fu K. Pedigree analysis of French Z Canadian Families with T14484C Leber's hereditary optic neuropathy // Neurology. - 1988. - Vol. 50. - P. 417-422. 22. Mashima Y., Kigasawa K., Hasegawa H. High incidence of pre-excitation syndrome in Japanese families with Leber's hereditary optic neuropathy // Clin. Genet. - 1996. - Vol. 50 (6). — P. 535-537. 23. Newman NJ, Lott MT, Wallace DC The clinical characteristics of pedigrees of Leber's hereditary optic neuropathy with the 11778 mutation // Am. J. Ophthaimol. - 1991. - Vol. 111. - P. 750-762. 24. Newman NJ Mitochondrial disease and the eye. Neuro-ophthalmology in systemic disease. - 1992. - R. 3. 25. Nikoskelainen EK, Marttila RJ, Huoponen K., Juvonen V. Leber's “plus”: neurological abnormalities in patients with Leber’s hereditary optic neuropathy // J. Neurol. Neurosurg. Psychiatry. - 1995. - Vol. 59(2). — P. 160-164. 26. Nikoskelainen EK, Savontaus M.-L., Huoponen K. Pre-excitation syndrome in Leber's hereditary optic neuroretinopathy // Lancet. - 1994. - Vol. 344 (8296). — P. 857-858. 27. Oostra RJ, Bolhuis PA, Wijburg FA Leber's hereditary optic neuropathy: correlations between mitochondrial genotype and visual outcome // J. Med. Genet. - 1994. - Vol. 31. - P. 280-286. 28. Ortiz RG, Newman NJ, Manoukian SV Optic disk cupping and electrographic abnormalities in an American pedigree with Leber's hereditary optic neuropathy // Am. J. Ophthalmol. - 1992. - Vol. 113. - P. 561-566. 29. Riordan-Eva P., Sanders MD, Govan GG The clinical features of Leber's hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation // Brain. - 1995. - Vol. 118(2). — P. 319-337. 30. Scott C. N. Oliver, Jeffrey L. Bennett. Genetic disorders and the optic nerve: a clinical survey // Ophthalmology Clinics of North America. - 2004. - Vol. 17(3). — P. 435-445. 31. Shaikh S., Ta C., Basham A., Mansour S. Leber hereditary optic neuropathy associated with antiretroviral therapy for human immunodeficiency virus infection // Am. J. Ophthalmol. - 2001. - Vol. 131. - P. 143-145. 32. Shoffner JM, Wallace DC Oxidative phosphorylation diseases. Disorders of two genomes // Adv. Hum. Genet. - 1990. - Vol. 19. - P. 267. 33. Smith JL, Hoyt WF, Susac JO Ocular fundus in acute Leber hereditary optic neuropathy // Arch. Ophthalmol. - 1973. - Vol. 90. - P. 349. 34. Vaphiades MS, Newman NJ Optic nerve enhancement on orbital magnetic resonance imaging in Leber's hereditary optic neuropathy // J. Neuroophthalmol. - 1999. - Vol. 19. - P. 238-239. 35. Wallace DC, Singh G., Lott MT, Hodge JA Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy // Science. - 1988. - Vol. 242. - P. 1427-1430. 36. Yeates FM Causes of binocular legal blindness in an Australian metropolitan community // Aust. J. Ophthalmol. - 1983. - Vol. 11. - P. 321-323.
Causes of Leber's hereditary optic neuropathy
The causes of Leber neuropathy, in which the optic nerve atrophies, are genetic. The disease, caused by a specific mutation in mitochondrial genes, is transmitted by women to their offspring. Men and women inherit pathology from their mother. Why is this happening? A huge amount of DNA is found in the cell nucleus and only a small part of it is in the mitochondria. Nuclear genes are inherited from both mother and father, but mitochondrial genes are inherited only from the mother. A man who inherits a mutation will not pass it on to his descendants.
In a significant proportion of people who are carriers of the mutated gene, the disease will not become apparent. More than 85% of women and 50% of men who are carriers of the LHON syndrome gene will not be affected by the disease. The reasons influencing the progression of the disease are not clear, but it is reliably known that the disease can be triggered by unfavorable environmental conditions, stress, infections, and the toxic effects of tobacco and alcohol.
Diagnosis of Leber optic neuropathy
Diagnosis of the disease is complicated by the similarity of its symptoms with ischemic neuropathy or with certain pathologies - for example, multiple sclerosis. It is also difficult to track the history, since not all carriers of the mutated gene develop Leber neuropathy. An extensive ophthalmological examination can only establish the presence of pathology, but to determine the cause, genetic diagnosis will be needed.
Features of diagnosing Leber hereditary optic neuropathy:
- general clinical examination;
- Mitochondrial DNA analysis;
- visual field examination;
- fundus examination;
- coherence tomography and electroretinography (necessary to exclude retinal pathologies).
Specialists are recommended to carry out a differential diagnosis of LHON syndrome by comparing symptoms and examination data with other diseases affecting the optic nerve. For example, with ischemic, toxic neuropathy. A complete ophthalmological examination shows that the optic disc is inflamed, and vascular telangiectasia (dilation of small capillaries) is also noticeable.
How does the disease manifest itself?
Leber optic nerve atrophy can be suspected based on several characteristic signs:
- A person's visual acuity sharply (sometimes gradually over a couple of years) decreases. A decrease in vision can immediately occur as a bilateral process, or first vision deteriorates in one eye, and then in the second eye.
- So-called scotomas—dark “blind” spots—may appear in the center of the visual field.
- The early stage of the disease is characterized by redness of the optic nerve disc. After a couple of weeks, his temporal half turns pale. This symptom can only be noticed during an ophthalmological examination.
- In approximately half of the cases, along with signs of eye damage, neurological manifestations of the disease can be observed - tremor, dizziness, deterioration of reflexes, headache, pronounced muscle tone.
Leber's hereditary optic neuropathy - can the disease be cured?
While hereditary optic neuropathy is considered an incurable disease, scientists are constantly searching for new ways to treat such a pathology. Thanks to gene therapy methods, it is possible to significantly slow down the progression of diseases and improve the quality of life of patients.
The rapid progression of Leber's disease leads to rapid loss of vision in people who have seen normally all their lives and had no vision problems. A disappointing diagnosis, as well as information that the disease has not yet been treated, seriously affects the well-being of such patients who were not prepared for disability. Many of the existing methods of treating pathology have proven to be ineffective, including surgical intervention.
Fortunately, scientists involved in rare pathologies are working hard to find a cure. Experts place particular hope in gene therapy, and many research groups have already achieved good results. Sometimes an obstacle to continuing experiments is their ethical side, since further research involves the use of the developed technique on humans. Are there any successes at least using experimental models? Yes, geneticists have already managed to find a way to solve the problem.
Thus, a group of scientists from Miami, using experimental models, proved that mutated genes can be safely replaced with healthy ones, this will prevent deterioration in the nutrition of optic nerve cells. To correct a genetic defect in the mitochondria, it is necessary to introduce normal DNA - this will correct the disorder and restore visual function. Scientists report that this approach will also be effective against other diseases caused by mitochondrial mutations, as well as various disorders associated with the aging process.
Causes
In most cases, transmission of the disease is recorded in an autosomal recessive manner. This means that vision pathology will be caused in a child if both parents have a defective gene.
Due to the fact that the cells of the pigment epithelium and photoreceptors in a newborn are not able to divide, Leber’s amaurosis manifests itself. Today, this disease is considered a heterogeneous group of disorders that cause the destruction of cones and rods.